Fig. 1
Assay development and validation plan
In vitro diagnostics (IVDs) for human use come under statutory regulations in most jurisdictions (e.g., the US Food and Drug Administration (FDA), the European Commission (EC)), and although veterinary IVDs are generally free of these controls, there is a responsibility, and usually a commercial necessity, to ensure that tests are fit for purpose. Both in-house and commercial assays (commercial kits used in modified form or for off-label applications fall within the in-house status) need to be backed by the same level of evidence supporting their performance against a specification.
Once an assay has been introduced, systematic quality assurance (QA) activities are essential to ensure that the assay continues to perform according to the requirements of the specification and fulfills its purpose.
Quality control (QC) procedures must also be applied to maintain the uniformity of the procedures and materials used to perform the assay. Output data monitoring using multi-rule criteria is a useful aspect of QC.
2 Validation of Molecular Diagnostics
This chapter refers to all nucleic acid-based (i.e., molecular) assays including those designed for diagnostic and reference purposes.
The World Organisation for Animal Health (OIE) has developed a register of diagnostic kits certified by the OIE as validated as fit for purpose [1]. Examples of the most common purposes are to:
1.
Demonstrate freedom from infection in a defined population (country/zone/compartment/herd): (1a) “free” with and/or without vaccination and (1b) reestablishment of freedom after outbreaks.
2.
Certify freedom from infection or presence of the agent in individual animals or products for trade/movement purposes.
3.
Eradication of disease or elimination of infection from defined populations.
4.
Confirmatory diagnosis of suspect or clinical cases (includes confirmation of positive screening test).
5.
Estimate prevalence of infection or exposure to facilitate risk analyses (surveys, herd health status, disease control measures).
6.
Determine immune status of individual animals or populations (post-vaccination).
These purposes are broadly inclusive of many narrower and more specific applications of assays. Such specific applications and their unique purposes need to be clearly defined within the context of a fully validated assay.
The term “validation” is often used very loosely and can cover a variety of different processes. From the manufacturing industry, “Validation is a quality assurance process of establishing evidence that provides a high degree of assurance that a product, service, or system accomplishes its intended requirements. This often involves acceptance of fitness for purpose with end users and other product stakeholders.” Validation is an evidence-based process that requires proper planning in order to ensure that newly developed assays comply with laboratory standard systems. There are a number of stages in this process, including planning and inception, assay development and optimization, assay validation, rollout and verification (i.e., a quality control process that is used to evaluate whether the assay complies with its specification), and finally implementation. Method validation can be used to judge the quality, reliability, and consistency of analytical results. It is therefore an integral part of any good analytical practice. Annex 15 to the European Union (EU) Guide to Good Manufacturing Practice [2] which deals with qualification and validation provides a useful context.
Analytical methods need to be validated before their introduction into routine use and revalidated whenever the conditions under which the original validation was done change (e.g., use of an instrument with different characteristics or samples within a different carrier matrix) and whenever the method is changed or modified beyond the original specification. The changes to a protocol that may be considered significant and that therefore require assay revalidation with adequate evidence for equivalent performance depend on the specific details of the test. Validation may be extensive, for example, in the case of a newly developed in-house assay, or narrow in scope, for example, in the case of a commercial assay already in use which has had minor modifications. Various situations are likely to arise in which it is appropriate to repeat only a subset of validation tasks. For example, if the extraction method is changed, it may not be necessary to carry out specificity checks, but the sensitivity will require reassessment. It is essential to provide documentary evidence that any assay is suitable for its intended purpose.
The laboratory may already have in-house or commercial assays in use for which no specific evidence of previously undertaken validation or verification is available. Almost certainly, this work would have been performed, but historically there may not have been a good culture of record keeping. It is important to provide documentary evidence of fitness for purpose. It is not normally necessary to repeat validation and verification work, and in practice, it may not be possible to retrospectively undertake this. It may be sufficient to prepare a file referring to existing evidence, such as results from interlaboratory comparisons or other studies undertaken, copies of published papers, internal quality control (IQC) and external quality assessment (EQA) results, etc. However, the process of reviewing validation data may highlight factors that require confirmation.
3 Quality Assurance
Quality assurance is the process whereby the quality of laboratory reports can be guaranteed and comprises all the different measures taken to ensure the reliability of investigations. It is not limited to the technical procedures performed in the laboratory. Therefore, although procedures within the laboratory that ensure that the testing procedures (the analytical phase) are reliable are important, consideration must also be given to the pre- and post-analytical phases, where the majority of the errors in the entire testing pathway occur.
The laboratory does not have control of many of the pre-analytical steps, such as what and how specimens were taken, labeling (of samples and request forms), and transportation. However, it can influence these activities by providing guidance in the form of a user manual. This provides details of those key factors related to the specimen which are known to affect the performance of the test or the interpretation of the results and instructions for transportation of samples, including any special handling needs. The laboratory should also have a procedure which defines the criteria for specimen rejection. It is good practice to notify the user concerning rejected specimens.
Many veterinary laboratories are accredited by the national accreditation body to the international standard ISO 17025 [3]. Accreditation is a means of assessing the technical competence of the laboratory and provides assurance to the laboratory and its customers that its service is fit for purpose.
The key elements of accreditation are competency of staff; use of documented, validated procedures; appropriate management and use of equipment and reagents; and a system of evaluation and quality improvement, involving internal audit, recording, and management of user complaints and nonconformities.
There is a much used saying among “quality” professionals, “If it isn’t written down it didn’t happen.” Good record keeping is essential, for audit purposes (internal and external), to aid identification of root causes when problems are identified (e.g., determining which reagents or instruments might be responsible for a poor performing test) and evidence of due diligence if the quality of testing is questioned by a customer.
The most important aspect of quality is the culture of the organization. Some organizations regard accreditation as a tick box exercise. For these, compliance with accreditation standards will always be a burden. The efforts required getting ready for assessment visits by the accreditation body, to update standard operating procedures overdue review, to carry out audits, and to close nonconformities that should have been dealt with weeks or months ago, become a last minute struggle. For such organizations, the quality management system (QMS) is in place to maintain accreditation and not to ensure best practice. Another well-known saying is that “Quality starts at the top.” If senior management recognize the value of the QMS, resource it appropriately, attend quality management meetings, and support quality related activities, then the culture of the organization will be enhanced as will be its performance.
4 Quality Control
Quality assurance of test methods will be provided by a combination of internal quality control (IQC), internal quality assessment (IQA), and external quality assessment (EQA).
4.1 Internal Quality Control
IQC is the analysis of material of known content in order to determine in real time if the procedures are performing within predetermined specifications. It is primarily the day-to-day monitoring of reproducibility or precision designed to detect errors in any single days analytical procedure. Performance of control material within predefined limits is essential for technical validation of a diagnostic test. The type of control used will depend on the type of assay. For qualitative assays, controls may just consist of a positive or negative sample. For quantitative assays, quality requirements of controls need to be determined for high and/or low clinical decision limits, depending on the analyte. A good understanding of the assay is essential in order to ensure that the appropriate controls are used. There is further discussion on the use of controls for polymerase chain reaction (PCR) assays later in this chapter. A number of papers have been published on the use and interpretation of controls in veterinary laboratories [4, 5].
Commercial assays will include assay controls but the laboratory should not rely on kit controls alone. Manufacturers adjust controls from batch to batch to give consistent results although assay sensitivity may vary between batches. This consistency is an obvious aid to the user, but it does mean that if you only use the controls that come with the kit, you will not detect any batch to batch variation. Therefore, it is recommended that the laboratory also use independent internal quality control materials, either purchased from a commercial source or prepared in-house. Use of the same internal control material over an extended period monitors batch to batch variation.
Internally prepared controls must:
Behave like real samples
Have sufficient to last for a period of time, ideally at least a year
Be stable over the period of use
Be appropriately apportioned for convenient use
Vary little in concentration between aliquots
Operate within the linear region of the assay
A series of QC results may be plotted as run charts, also known as Levey Jennings or Shewhart charts. These show the values plotted against the mean and usually the 1, 2, and 3 standard deviation (SD) values. Westgard rules [6, 7] may then be used to define specific performance limits and detect both random and systematic errors. Three of the six commonly used Westgard rules are warning rules, the violation of which should trigger a review of test procedures, equipment calibration, and reagent performance. Three are mandatory rules which, if broken, should result in the rejection of results in that assay run.
It is important to note that a system of quality control is unlikely to improve a method that is fundamentally unsound. Performing quality control does not by itself improve the quality of any assay. It is the interpretation of the data obtained and appropriate actions taken that will lead to quality improvement.
4.2 Internal Quality Assessment (IQA)
IQA is the repeat testing of a percentage (typically 0.5–1 % of workload) of routine test samples to determine the laboratory’s ability to obtain reproducible results. IQA is a commonly used tool in clinical microbiology laboratories but less so in other disciplines. Consistency is an important measure of quality assurance, so repeat tests continually giving the same results as the original show a system that is in control. The fact that IQA is performed on so many samples throughout the year means that the results obtained are statistically significant.
A specimen is split in two on arrival into the laboratory, and while one part is put through the test procedure in the normal way, the other one is given an IQA number and a new request form is created. The IQA sample is then tested and reported in the normal way, except that the report is sent to a member of staff rather than a requesting clinician. The IQA result is then compared with the original result and any discrepancies noted. Discrepancies are normally classed as either minor or major. Minor ones show variation but would not affect the result, whereas major discrepancies are likely to lead to a different result. All discrepancies should be investigated and major ones are likely to require repeat testing and may result in an amended report being issued.
IQA requires the repeat sample to be booked in, for tests to be selected and then performed, results to be validated, and a report to be produced, with interpretive comments included. Therefore, IQA assesses important aspects of the pre- and post-analytical phase which take place within the laboratory as well as the analytical phase itself. Although on its own, IQA does not prove that the laboratory is actually getting the right result, it is a good test of the system and, when combined with other quality control measures, is helpful in evaluating process control.
4.3 External Quality Assessment (EQA)
External quality assessment is usually an externally organized function that monitors the efficacy of quality assurance procedures. It compares the performance of different testing sites by allowing the analysis of an identical specimen at many laboratories, followed by comparison of individual results with those of other sites and with the correct answer. EQA acts as a check on the efficacy of internal quality control procedures.
EQA is performed on a limited number of samples, and the process is inevitably retrospective, providing an assessment of performance rather than a true control for each test performed. It gives participants an insight into their routine performance so that they can take action to achieve improvements. EQA is an educational tool. To achieve the greatest benefit from EQA, samples must be treated the same way as routine samples.
The diagram in Fig. 2 is a good illustration of how to interpret QC results in terms of assay performance. The correct EQA result could be the dart closest to the bull’s eye in the first dartboard on the left. Therefore, the correct EQA result does not show that the laboratory is capable of getting consistent results or of getting the correct result consistently. If IQA results are consistently the same as the original test results, they show good precision as shown in dart boards 2 and 3 but do not distinguish between these two options, i.e., show good process control but not whether the assay is good. If IQC results are consistently accurate (mean close to expected value of the control and small SD values), then this shows good precision and a good assay, as indicated by dartboard 3. However, only the examination phase is assessed by IQC.
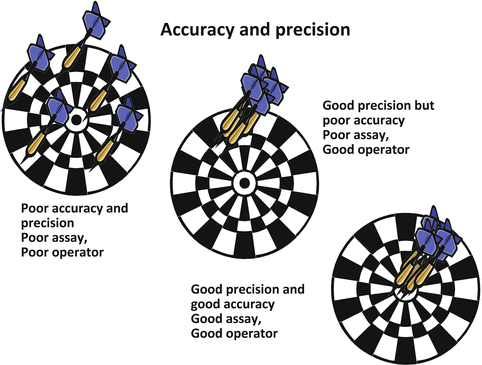
Fig. 2
Illustration of how to interpret quality control results in terms of assay performance
Clearly, EQA, IQA, and IQC all have their role to play. EQA allows comparison with results obtained by all other laboratories participating in the scheme. IQA shows whether or not the laboratory has the pre-examination, examination, and post-examination activities in control, and IQC shows whether or not the actual results are correct. By performing EQA, IQA, and IQC and reviewing the data obtained, you get a powerful indication whether or not you have a well-controlled process (and competent operator) and also a good assay.
4.4 Personnel
All personnel involved with the validation, quality assurance, and quality control of diagnostic tests must have clearly defined lines of accountability and be equipped with appropriate knowledge, competency, and experience. Records of their qualifications must be available.
4.5 Equipment
All equipment used in the assay validation exercise must be maintained, serviced, calibrated, and monitored as appropriate to ensure that it is suitable for use. This is essential to ensure that all conditions can be reproduced accurately during subsequent routine production of reagents and performance of the assay.
5 Planning and Inception
5.1 Establishment of the Project
The drivers for introduction of new diagnostics are most commonly gaps in capability and capacity or opportunities for improved service presented by new knowledge and technology. During the planning phase, the aim is to produce a clear, agreed project plan. A vital part of the process is to ensure that the project is properly resourced.
5.2 Establishment of a Review Team
A suitable panel (“review team”) should then be established to review the plan. This panel will also review the progress of the validation work, evidence that the test is fit for purpose and that plans for monitoring of test performance are in place. It is important to note that the size and composition will depend on the complexity of the project. The principle of having a review team will apply however simple the project. However, a straightforward revalidation following a minor change may be undertaken and overseen by just one person. However, final sign-off of the project must be undertaken by an appropriate senior member of staff.
The project leader and project manager roles may be performed by a single individual. The project manager, who has overall responsibility for the completion of the validation project and responsibility for signing off the completed validation file and the documented standard operating procedure (SOP) for performance of the assay, should, most appropriately, be at least at team leader level (however described). The project leader has responsibility for the project work, including laboratory activity needed for the validation, data analysis, compilation of the validation file, report writing, presentation of data to review meetings, writing and maintaining the SOP, and training of staff to carry out the procedure described in the new SOP.
One member of the review team should be responsible for ensuring that all documents relating to the project (i.e., the contents of the project dossier) are brought to the attention of senior management. This person must also ensure that the management are informed of the key project events (i.e., project inception and funding, development work successful in meeting design requirements, project abandonment, validation study completion, declaration that the test is fit for purpose and plans for deployment). An example of a suitable team would be two laboratory scientists, at least one clinical representative and the local quality representative. It is highly desirable that one or more of the members of the team should be a potential end user of the assay, as this will be useful in providing input to the validation parameters to ensure clinical utility. It is recommended that at least one of the members of the team has statistical expertise, but alternatively a statistician should be consulted to give advice on the validation study design. The project manager or project leader, but preferably not both, unless both roles are held by a single person, may be members of the review team.
The review team has the following responsibilities:
1.
To assess how the assay will improve or fill gaps in the current testing repertoire. This should include identification of the diagnostic need, the currently available alternatives, the end users, and any other stakeholders.
2.
To compile a register of risks associated with project success or failure and implementation of the assay (e.g., users might use inappropriate specimen types or misinterpret the results). The register should specify design actions to be taken in mitigation of the specified risks.
3.
To ensure that laboratory safety issues associated with the test and the validation program have been assessed.
4.
To ensure that the engagement of collaborative partners to provide expertise or share costs has been considered.
5.
To ensure that training and other human resources issues are addressed.
6.
To ensure that the means for efficient project management have been established, including the nomination of advisors and reviewers as necessary.
7.
To approve the assay validation plan and then, when complete, to review the validation study data and decide whether the assay is suitable for deployment.
8.
To review the assay deployment and post-deployment plans.
9.
To ensure that the project dossier is maintained.
6 Assay Validation Plan
The objective of the assay validation plan is to ensure that the assay conforms to the required specification. The specification may be broad, for example, requiring that the assay accurately and reliably measures only the analyte of interest in clinical samples with the required level of sensitivity. Ensuring test reliability will include the need to control the uniformity of the assay procedure and reagents over time and the maintenance of full result traceability.
The review panel and project leader should conduct planning meetings, perform literature searches, appraise options, and, where possible, consult with other centers that carry out the same or similar assays. The project plan documentation should be version controlled. When the project plan has been agreed, the project leader will be responsible for performance of the laboratory tasks. When the agreed project milestones have been reached, a review panel meeting should be held with the project leader presenting the data. Any follow-up work and/or analysis, if necessary, will be agreed, following which the project lead will generate the technical report. The technical report is then circulated to the review panel, which will then either sign off the assay as ready for rollout for routine use (technical transfer including training of routine diagnostic staff) or request further work that is carried out before the assay can be signed off as suitable.
The technological details of the proposed assay, including information on the platforms, reagents, controls, and protocol to be used, should be specified in the validation plan. Sample preparation methods form an integral part of the diagnostic test for validation purposes.
The plan should specify the sample types to be evaluated and both the essential (i.e., minimum) and optimal sample volumes.
7 Validation Design
The project leader should prepare the validation plan including the following points that are based on the STARD initiative [8] and external literature including MIQE [9]:
1.
Define the purpose and objectives of the validation study. For example, the study may be intended to validate the performance of a new assay or may aim to demonstrate that a significantly modified assay or protocol variant gives results within the tolerance of the original.
2.
Identify any training requirements to ensure everyone involved in the validation has suitable levels of competency. Ensure training records are up to date for procedures being carried out.
3.
Identify any risk assessments which need to be reviewed or written.
4.
Identify the available standards or reference materials. These act as controls to allow the assay to be standardized, facilitate assay comparison, and permit stability of the assay to be determined over time.
5.
Identify the assay to be used for comparison with the assay undergoing validation. This should be the currently accepted “gold standard” where one is available.
6.
Design an analytical validation study to test the sensitivity and specificity of the assay using control materials, extracts from a wide range of strains/variants of the target organism, specimens spiked with the target organisms, and a range of unrelated strains or species that could be present in a sample, but that should not give a positive result.
7.
Design a clinical validation study appropriate to the clinical context (e.g., surveillance, screening, clinical diagnosis). Choose the study group including species, case definitions, inclusion/exclusion criteria, and study settings.
8.
Identify the types (i.e., specimen, method of sampling, transport and processing) and numbers of samples to be tested. Consider the need to include known positives, known negatives, low and high positives, and samples which are known or likely to be problematic (e.g., containing inhibitors or possibly cross-reactive markers).
9.
Select appropriate statistical tools for determination of an appropriate sample size and to avoid bias. It is essential to consider statistical requirements to ensure that results are statistically significant. The sample size needed to ensure statistically significant results depends on a number of variables. Some guidance is given in Tables 1 and 2. The validation design should avoid discrepant analysis bias. Some samples may give discordant results with the new test compared to a “gold standard.” If only these samples are retested, bias is introduced as there is a probability that the second analysis will give concordant results for some of these samples. Both concordant and discrepant samples should be retested to avoid bias.
Table 1
Relationship between sample size and 95 % confidence interval
Number of infected (noninfected) subjects requireda | Estimated test sensitivity (or specificity)b | |||||
|---|---|---|---|---|---|---|
50 % | 60 % | 70 % | 80 % | 90 % | 95 % | |
50 | 13.9 % | 13.6 % | 12.7 % | 11.1 %
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

| ||