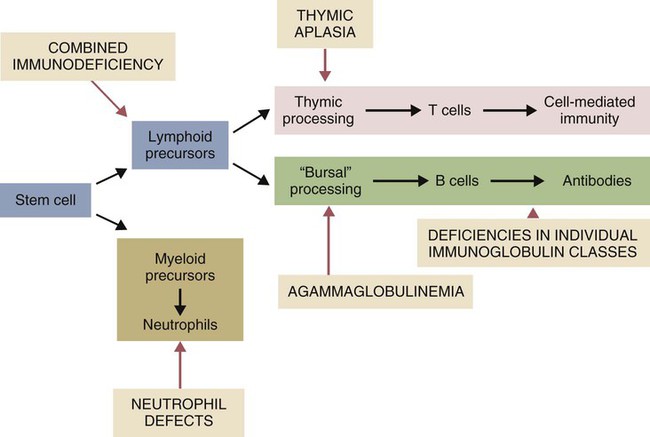
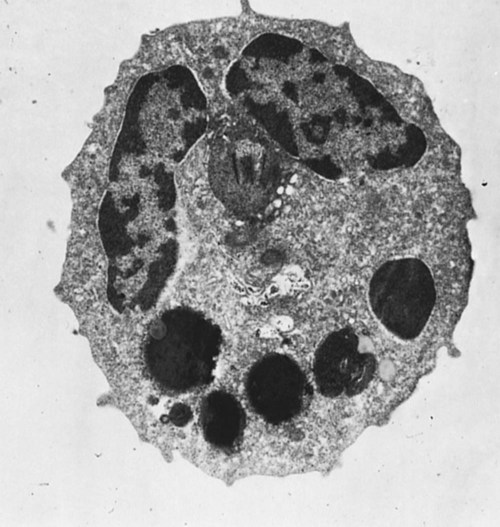
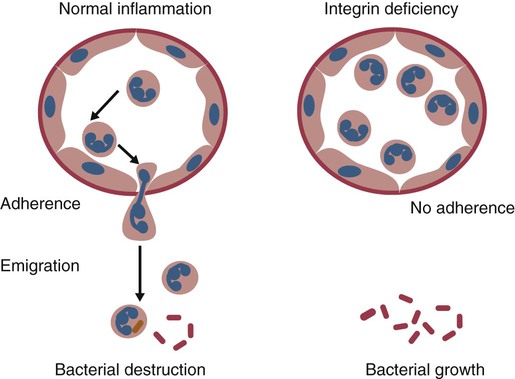
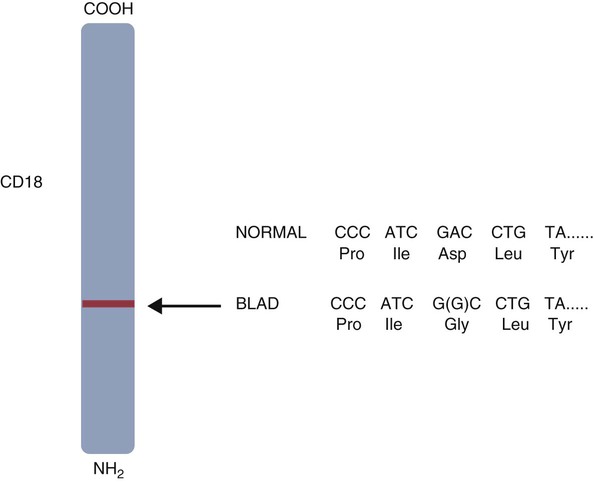
• As a result of genetic mutations, defects may develop in the developing immune system, resulting in immunodeficiency in newborn animals. • Many different inherited defects have now been identified in domestic animals, especially in inbred breeds in which heterozygosity is reduced. • Defects in innate immunity include deficiencies in phagocytosis, leukocyte adherence, and intracellular killing, leading to increased susceptibility to bacterial diseases. • Defects in T cell function generally predispose an animal to overwhelming virus infections. • Defects in B cell function and immunoglobulin production predispose animals to overwhelming bacterial disease. • Combined immunodeficiencies are most severe since affected animals lack resistance to all infectious agents. Inherited deficiencies in innate immunity include defects in the various stages of phagocytosis as well as the complement deficiencies described previously (Chapter 7). Phagocytic defects are well recognized in domestic animals. Chédiak-Higashi syndrome is an inherited disease of Hereford, Japanese black, and Brangus cattle, Aleutian mink, blue smoke Persian cats, white tigers, beige (bg/bg) mice, Orca whales, and humans. It is an autosomal recessive disease resulting from a mutation in a gene (LYST) that encodes a protein that controls lysosomal membrane fusion. The LYST gene is found on bovine chromosome 28. In Chédiak-Higashi cattle, there is a missense A : T → G : C mutation that results in replacement of a histidine with an arginine residue. The defect produces abnormally large secretory lysosomes in neutrophils, monocytes, eosinophils, and pigment cells (Figure 37-1). The enlarged neutrophil granules result from the fusion of primary and secondary granules. The leukocyte granules of affected animals are more fragile than those of normal animals, rupturing spontaneously and causing tissue damage, such as cataracts in the eye. These leukocytes have defective chemotactic responsiveness, reduced motility, and reduced intracellular killing. Cytotoxic T cells fail to excrete their granzyme-rich lysosomes. In order for neutrophils to leave inflamed blood vessels, they must first bind to vascular endothelium. This adhesion is mediated by neutrophil integrins. In the absence of these integrins, neutrophils cannot bind to endothelial cells and are unable to emigrate into tissues (Figure 37-2). Thus bacteria in tissues can grow freely without fear of attack by neutrophils. Canine leukocyte adhesion deficiency (CLAD) results from a defect in the integrin CD11b/CD18 (Mac-1). In Mac-1-deficient dogs, neutrophils cannot respond to chemoattractants, trap complement-coated bacteria (Mac-1 is a complement receptor), or bind to endothelial cells. Affected dogs suffer recurrent infections, despite the fact that their blood neutrophils are greatly elevated. BLAD results from a point mutation in the gene coding for CD18 (Figure 37-3). As a result, an aspartic acid residue is replaced by a glycine, and functional CD18 is not produced. In the absence of this chain, complete integrins cannot be assembled. Neutrophils fail to attach to vascular endothelial cells and cannot emigrate from blood vessels. Healthy carriers have a single copy of the mutated gene and thus have abnormally low levels of CD18 (Figure 37-4). Through the use of a PCR test, the presence of the altered gene can be demonstrated. In this way it has been shown that one bull, Osborndale Ivanhoe, with thousands of registered sons and daughters, was a carrier of this gene. As a result, the defective gene was widespread and common among Holstein cattle in the United States (14% of bulls, 5.8% of cows). Fortunately, carrier animals can be rapidly detected and removed from breeding programs. The inherited immunological defects have served to confirm the overall arrangement of the immune system, as outlined in Figure 37-5. For example, if both the cell- and antibody-mediated immune responses are defective, it may be assumed that the genetic lesion operates at a point before thymic and bursal cell processing—that is, a stem cell lesion. A defect that occurs only in thymic development is reflected in an inability to mount cell-mediated immune responses, although antibody production may be normal. Similarly, a lesion restricted to B cells is reflected by impaired antibody responses.
Primary Immunodeficiencies
Inherited Defects in Innate Immunity
Chédiak-Higashi Syndrome
Canine Leukocyte Adhesion Deficiency
Bovine Leukocyte Adhesion Deficiency
Inherited Defects in the Adaptive Immune System
Primary Immunodeficiencies
Only gold members can continue reading. Log In or Register to continue

Full access? Get Clinical Tree


