, Christine Van Broeckhoven1 and Samir Kumar-Singh1
(1)
Neurodegenerative Brain Diseases Group, VIB Department of Molecular Genetics and Laboratory of Neurogenetics, Institute Born-Bunge, University of Antwerp, Wilrijk, Belgium
(2)
Department of Histology, University of Medicine and Pharmacy of Craiova, Craiova, Romania
Abstract
Alzheimer’s disease (AD) and frontotemporal dementia (FTD) are two most common forms of presenile dementia where insoluble protein deposits as intra- or extracellular aggregates. During the past decade, a number of mouse models have been devised based on human mutant genes associated with familial forms of disease. Partly due to such experimental models, enormous progress has been made in the understanding of mechanisms by which amyloid-β or tau protein is toxic to neurons and causes part of the cognitive/behavioral or neuropathological features characteristic of AD or FTD. This chapter enumerates transgenic mouse models commonly used in AD and FTD research and discusses how these have served as an important research tool in defining critical disease-related mechanisms. Furthermore, this chapter also summarizes how these mouse models have contributed in identification of potential drug targets or in the evaluation of novel therapeutic approaches in delaying the onset or progression of these devastating diseases.
Key words
Transgenic mouse modelsAlzheimer’s diseasefrontotemporal dementiafrontotemporal lobar degenerationAβtauneuropathologybehaviortherapy1 Introduction
Alzheimer’s disease (AD) is the most frequent form of neurodegenerative disease resulting in progressive loss of memory and cognitive abilities. It is responsible for the cognitive decline in up to three quarters of all dementia patients (1). It was not until the turn of the last century that this syndrome was recognized as an individual entity and its two pathological hallmarks, the amyloid plaques and the tau neurofibrillary tangles became widely accepted (2). However, the precise connections between amyloid precursor protein (APP) and tau proteinopathies as well as any relation of these proteinopathies to synaptic and neuronal loss in AD are still largely unknown. The identification of the early onset, familial AD (FAD)-linked APP (3), and presenilin 1 and 2 mutations (4,5) offered for the first time a possibility to model the disease, and thus opened new frontiers in AD research. Several overexpression transgenic mouse models were made that hosted different pathogenic mutations that subsequently have been extensively characterized regarding the transcriptional promoter, the earliest time at which amyloid deposition appeared, the predominant type of amyloid-β (Aβ) deposition, and the associated neuropathological, biochemical, and behavioral changes. A decade of such investigations has shown that many of these mouse models have a number of similarities with human disease, thereby helping to gain further insights into different pathological aspects of the human disease. Thus, not surprisingly, many of these mouse models are also being utilized in current preclinical drug trials.
This book chapter, after addressing to some of the salient features of AD and amyloid metabolism, will discuss some common mouse models of AD and show how such mouse models have been instrumental in understanding the etiopathogenesis of Aβ amyloidosis. Such protein misfolding pathology (or proteinopathy) is also a common feature of other dementias. Of relevance to AD is also tauopathy, a co-proteinopathy in AD but a primary proteinopathy in frontotemporal dementia (FTD) of the tau type. We will therefore also discuss mouse models of tau and see how these have helped in finding common links between Aβ and tau pathologies.
2 APP and MAPT
2.1 APP and Aβ peptide
Amyloid precursor protein is a ubiquitously expressed single-transmembrane protein with a 590–680 amino acids (aa.) long extracellular amino terminal domain and a short cytoplasmic tail, which contains intracellular trafficking signals (Fig. 1) (6). APP transcripts undergo alternative splicing to yield eight possible isoforms of which APP-695, 751, and 770 isoforms predominate in the brain, with APP-695 isoform being mainly produced by neurons (7). Thus, most of the transgenic mouse and cellular models are based on APP-695 isoform, while some of the constructs also utilize APP-770 isoform.
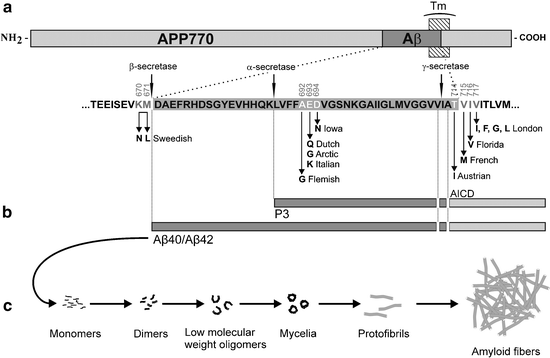
Fig. 1.
Schematic diagram of the amyloid precursor protein (APP) with pathogenic mutations and metabolic processing.
(a) The Aβ fragment is located between residues 671 and 714 and partially overlaps with the transmembrane segment (TM). The amino acid sequence of Aβ fragment (numbering accordingly with the APP770 isoform) is enlarged below and shows common pathogenic amino acid substitutions. Sites of α-, β-, and γ-secretase cleavage points are indicated by arrows. (b) The constitutive proteolytic cleavage by α- and γ-secretases leads to the formation of the short p3 peptide, and the alternative pathway leads to the formation of Aβ peptide, in both cases with a consecutive release of a C-terminal APP intracytoplasmic domain (AICD). The variations at the γ-secretase site cleavage lead to the formation of Aβ40/Aβ42 isoforms. (c) Various forms Aβ isoforms described in human pathology and transgenic mouse models.
While APP is normally processed by α- and γ-secretases to release a secreted APPα fragment (APPsα), a 3 kD p3 peptide and a 7 kD APP intracellular domain (AICD), the alternative processing of APP by β- and γ-secretases results in full-length Aβ, an N-terminal soluble APPsβ fragment, and the C-terminal AICD tail (8). The α- and β-secretases have recently been identified as members of the ADAM and BACE family of enzymes, respectively, and γ-site processing was found to be dependent on a pentameric complex in which presenilin 1 and 2 (PS) act as aspartyl transmembrane proteases (9,10). It is important to note that while these pathways were called amyloidogenic and nonamyloidogenic pathways, recent studies suggest that p3 or other N-terminally cleaved Aβ fragments are also deposited as amyloid (11) (see later). APP is processed in both the constitutive and alternative secretory pathways in the trans-Golgi network (6) and via the early endosomal-lysosomal-exosomal pathway (6,12). In neurons, APP and its catabolites are also trafficked along the microtubules within the axons via a kinesin-mediated mechanism and APP is observed to be present in synapses (13). This fits well with the arguable physiological function of APP in neurite growth and memory (14). In support of this proposed function, APP deficient mice show a moderate atrophy of corpus callosum together with astrogliosis but without any increase in morbidity or mortality (15). The anterograde transport of APP also might explain why some of the Aβ deposits in the dentate gyrus appear to originate from nerve terminals whose axons traverse the perforant pathway, and why lesions of perforant pathway in animal models of AD result in a reduction in hippocampal amyloid burden (16,17).
Aβ has two major C-terminal isoforms, Aβ40 and Aβ42. Aβ40 comprises 90–95% of secreted Aβ and is the predominant species found in the cerebrospinal fluid while less than 10% of the secreted Aβ is Aβ42 (18). The secreted fraction of Aβ presents as monomers/dimers and soluble Aβ oligomers, which are passively redistributed in brain parenchyma and cleared (19). Several Aβ clearance mechanisms have been described. First, Aβ is taken up and degraded by microglial and astroglial cells (20). In addition, Aβ is also degraded by proteases including neprilysin (NEP), insulin-degrading enzyme (IDE), endothelin-converting enzymes-1 and -2 (ECE), matrix metalloproteinase-2 and -9 (MMPs), and tissue plasminogen activator (tPA) (21). Of these, NEP, IDE, and ECE1 are active intracellularly while IDE, tPA, and MMPs act at the cell surface or are secreted and activated in the interstitial fluid (ISF) (21). Knockout models for many of these proteases show a significant elevation of murine brain Aβ, while their overexpression counterparts show a reduction in Aβ levels and retardation of plaque formation (22). Human studies also show that a local reduction in expression of some of these proteases correlates with a high plaque burden (23) (Fig. 3, upper panel). Lastly, Aβ is also drained out from the brain parenchyma by two mechanisms involving blood vessels. One of these mechanisms is a direct Aβ transport across the blood-brain barrier (BBB) mediated by LDL receptor-related proteins-1 and -2 (LRP-1, LRP-2), and regulated by α2-macroglobulin, apolipoprotein E (ApoE), and apolipoprotein J (ApoJ), respectively (24) (see later). This direct vascular clearance mechanism is highly efficient in clearing not only physiological Aβ but also pathological amounts of Aβ in transgenic mouse models (25). It is postulated that if the influx of circulating Aβ would be completely stopped, LRP-mediated transport could remove the entire Aβ pool from brain ISF within less than 1 min under physiological conditions, or in approximately 40 min at pathological levels (26). A reduction of LRP-1 expression has indeed been observed in AD patients as well as in mouse models (27). The second vessel-related Aβ clearance route utilizes the periarterial ISF drainage pathway where Aβ in ISF is collected around periarterial space (also known as the Virchow-Robbin space) that reaches the cerebrospinal fluid (CSF) in the subarachnoid space. From here, along the perivascular spaces of the circle of Willis, the fluid reaches the olfactory bulbs, then passes with perforant lymphatic channels through the cribriform plate to nasal submucosa and finally to the cervical lymph nodes (28). Tracer experiments in animal brain demonstrate that this is equivalent to lymphatics of brain (29). The ISF drainage pathway constitutes a major route of Aβ elimination in situations when BBB becomes less efficient (as in aging) or is overwhelmed (as in mouse AD models) (24,30). All mechanisms taken together, Aβ clearance from human brain normally exceeds its production with Aβ fractional production and clearance rates measured as 7.6% and 8.3% per hour, respectively (31).
Both soluble and fibrillar aggregates of Aβ are shown to be toxic and mouse models have been very useful in confirming the toxic potential of different Aβ aggregation states, either by showing inhibition of hippocampal long-term potentiation (LTP) or by demonstrating memory impairment after injecting aggregated Aβ species in rodent brain. For instance, together with human and in vitro studies, mouse models have been instrumental in showing that fibrillar and/or nonfibrillar forms of Aβ deposited in vessel walls are associated with degeneration of endothelial cells, smooth muscle cells (SMCs), and brain pericytes (32–34). Similarly, reduction in synaptophysin levels and disruption of neuronal calcium homeostasis has been shown in mouse models, suggesting a considerable role of soluble Aβ aggregates (35–37). Increasing data on varied models also indicate that Aβ can in turn activate microglia and thus indirectly damage neurons, contributing to the neurodegenerative process and cognitive loss (38,39). It is maybe for this reason that the “neuritic plaques” are more active as these are shown to be in histological proximity with inflammatory markers (40,41). Some of these issues will be discussed in relevant sections of this chapter.
2.2 MAPT and Tau
AD also shows tau proteinopathy with aggregated forms of hyperphosphorylated tau deposited in neurons as neurofibrillary tangles (NFTs) and neuropil threads. Hyperphosphorylated tau is also a major constituent of Pick bodies in FTD. Tau is a highly conserved protein that belongs to the family of microtubule associated proteins (MAPs) and is essential for the assembly of tubulin into microtubules (42). In human CNS, tau is abundantly expressed in the axons of mature and growing neurons; however, low levels of tau are also present in oligodendrocytes and astrocytes (43,44). The human tau gene (MAPT) is located on chromosome 17q21 and encodes 15 exons of which three exons (4A, 6, and 8) are never present in any mRNA expressed in human brain (reviewed in (45)). Tau mRNA is spliced in to six possible isoforms with lengths between 352 and 441 aa (46). These isoforms occur by combining two possible inserts (0N, 1N, 2N; encoded by exons 2 and 3) in the N-terminal domain, and 3 or 4 possible repeats in the C-terminal tail (3R or 4R, depending on the alternative splicing of exon 10) (45). Also, the six tau isoforms are not equally expressed in neurons, for example tau mRNA containing exon 10 is not found in granular cells of the dentate gyrus. The four repeat domains represents the microtubule-interacting domain, while the C-terminal acidic region projects between the microtubules, acting as a linker with the other components of the cytoskeleton and as a spacer for the other surrounding microtubules. In fact, tau proteins may interconnect microtubules with other cytoskeletal components such as neurofilaments and actin filaments. In mice lacking the tau gene, an increase in the microtubule associated protein 1A is observed, and this may compensate for the loss of tau functions (47). However, in these mice, axonal diameter in some neurons is particularly affected.
Phosphorylation is by far the most common posttranslational modification of tau. At least 25 different tau phosphorylation sites have been identified. Tau phosphorylation might have a physiological role, as during development fetal tau is more heavily phosphorylated than adult tau (48). Tau hyperphosphorylation and aggregation is also a major molecular change leading to accumulation of insoluble intracellular paired helical or straight filaments (PHF/SF) in a number of tau proteopathies such as AD, Pick’s disease (PiD), progressive supranuclear palsy (PSP), and corticobasal degeneration (CBD) (49). In the following sections, we will discuss how Aβ and tau interact in relevant mouse models.
3 Genetics of AD and FTD
Mutations in three genes have been deemed causal for autosomal dominant forms of AD: the APP gene (3), the PS1 gene (4), and the PS2 gene (5,50). Furthermore, while advancing age is the strongest risk factor to developing AD, different ApoE alleles are also shown to modulate the disease progression (51).
3.1 APP Mutations
Characterization of vascular amyloid in AD and Down’s syndrome patients led to the identification of the Aβ peptide and APP gene (52), which was followed by the identification of Aβ in parenchymal plaques (53). The first pathogenic mutation (E693Q) was also identified in a form of vascular amyloidosis called hereditary cerebral hemorrhages with amyloidosis – Dutch-type (HCHWA-D) (54,55). HCHWA-D patients are characterized by recurrent hemorrhages and extensive Aβ deposition in cerebral blood vessel walls, in the absence of senile plaques and tau-related pathology (56–58). Besides the occurrence of large lobar intracerebral hemorrhage, HCHWA-D patients may show cognitive deterioration, frequently associated with white matter abnormalities on MRI and small ischemic and hemorrhagic infarctions on pathological examination (59). At the level of APP processing, this mutation drastically increases the Aβ40/Aβ42 ratio, with decreased Aβ42 levels, both in vivo and in vitro (60,61). Moreover, the mutated Aβ Dutch peptides show an enhanced fibrillogenesis and toxicity towards vascular cells in various in vitro experimental setups, explaining to some extent its affinity to blood vessels (62,63).
The Flemish (A692G) APP mutation, identified subsequently, leads to both cerebral hemorrhages and AD (64). Pathologically, Flemish APP mutation carriers show senile plaques with the largest central dense-cores observed in AD and a severe degree of cerebral amyloid angiopathy (CAA) (41,65,66). Also, in contrast to HCHWA-D and consistent with a clinical diagnosis of AD, Flemish APP patients have considerable neurofibrillary pathology (41,65). Besides Dutch and Flemish APP mutations, other mutations that have been identified near the α-secretase processing site are Arctic (E693G), Italian (E693K), and Iowa (D694N) mutations (Fig. 1). These APP mutations lead to very similar phenotypes, dominated by amyloid angiopathy, diffuse amyloid deposits, and occasionally ischemic infarctions (67–69). This is explained by the fact that these α-secretase site APP mutations change the primary Aβ sequence and increase its vascular affinity and/or fibrillogenic potential including formation of Aβ oligomers.
The α-secretase site APP pathology contrasts sharply with pathology caused by mutations identified near the β- or γ-secretase sites. The only APP mutation near the β-secretase site is the double Swedish (K670N/M671L) mutation (70) that promotes β-secretase activity and increases the total Aβ production without altering the Aβ40/Aβ42 ratio (71). This is consistent with the pathological data in APP Swedish carriers, which deposit more Aβ40 compared to carriers of APP mutations near the γ-secretase site (72). The first γ-secretase site APP mutation identified was the London (V717I) APP mutation that results in an increased Aβ42/Aβ40 ratio, without altering total secreted Aβ (73,74). The enhanced secretion of more aggregatable Aβ42 directs the pathology towards a diffuse amyloid distribution predominantly constituted of Aβ42, and exceeding the burden observed in sporadic AD patients (72). Many γ-secretase site APP mutations have been identified and while almost all of these mutations decrease the total Aβ production and increase Aβ42/Aβ40 ratios (75), the pathology caused by Austrian (T714I) APP mutation is the most remarkable one. This mutation leads to a very aggressive form of AD with a very young age of onset and a diffuse, nonneuritic amyloid pathology (76). In vitro cell culture experiments in neuronal and nonneuronal cells showed that APP Austrian mutation led to one of the most drastic increases in the Aβ42/Aβ40 ratio in a panel of γ-secretase site mutations studied (76,77). Lastly, several independent duplications of the APP locus have been recently identified in French and Dutch families with autosomal dominant AD and/or lobar cerebral hemorrhage with prominent CAA (78,79). Similarly, Down’s syndrome patients, also carrying three copies of APP, develop CAA as young as 30 years and the severity of CAA and vessel-related risks increase with age (80). APP gene dosage should be considered as a situation similar to APP Swedish mutation because it also increases total Aβ without changing the Aβ42/Aβ40 ratio. These data underpin the growing consensus that increase in absolute levels of Aβ40 leads to a predominant vascular amyloidosis (81).
3.2 Presenilin Mutations
FAD mutations in presenilin 1 and 2 are more common than APP mutations (4,5,50). Structurally, PS are multiple transmembrane domain proteins localized in the endoplasmic reticulum, Golgi apparatus, and lysosomes as a part of the γ-secretase complex (6,82). The functions of PS are not entirely clear; however, PS1 knock-out mice die ante partum or immediately after birth and show lissencephaly, severe altered somitogenesis, skeletal, and vascular disturbances resembling a Notch phenotype (83,84). PS mutation carriers present with an earlier age of onset than APP mutation carriers, and with abundant diffuse-like amyloid pathology, perhaps because these mutations increase the production of the more insoluble and readily depositable Aβ42 isoform (75). Interestingly, most of the PS mutations tested in vitro not only increase Aβ42, but also lower Aβ40 production drastically with an overall reduction of total Aβ production (75,85). This might have important implications for amyloidogenic mouse models based on PS mutations (see further). Also, interestingly, select AD-linked PS1 mutations and the Volga-German PS2 N141I mutation associate with a marked CAA (86,87). While the precise cause is again unknown, it is plausible that PS mutations associated with predominant CAA favor the production of more soluble Aβ40 that is drained and deposited along the perivascular space in association with Aβ42 nidi (81).
3.3 Tau Mutations
As previously mentioned, the deposition of hyperphosphorylated tau as insoluble filaments in the brain is a pathological hallmark of several neurodegenerative disorders, defined as tauopathies. An autosomal-dominant inherited form of frontotemporal dementia with parkinsonism (FTDP) was initially linked to chromosome 17q21-22 in 1994 (88). In the following years, 13 additional families with FTD and parkinsonism with linkage to 17q21-22 were identified (45). These patients presented with disinhibition, loss of initiative, obsessive-compulsive behavior, and/or psychosis, followed by cognitive decline. In most patients, extrapyramidal symptoms occurred only late in the clinical course, but considerable heterogeneity is observed both between mutations and within families with the same mutation (89). In 1997 at a consensus conference, the term of frontotemporal dementia with parkinsonism linked to chromosome 17 (FTDP-17) was introduced to describe these patients (89) and in the following year mutations in MAPT were reported in the majority of these families (90,91). At present, 39 mutations in MAPT have been identified in 115 families, including missense mutations, silent mutations, in-frame single codon deletions, and intronic mutations (45). Most FTDP-17 MAPT mutations result in increased 4R/3R tau ratios, and modify tau splicing pattern or cause single aa changes, which probably result in impaired microtubule binding capacity or increased aggregability (45). On gross neuropathology, FTDP-17 patients present with a severe atrophy of the frontal and temporal lobes (or blade-edge atrophy described in classical Pick’s disease), although the superior part of the precentral gyrus is commonly spared. On microscopy, astrocytic gliosis and a massive neuronal loss are found in the superficial cortical layers and sometimes also in basal ganglia (92). Filamentous tau intracytoplasmic inclusions in neuronal and glial cells are the characteristic finding. Extensive mutation analysis revealed many other MAPT mutations in several additional FTDP-17 families, as well as in unrelated FTD, CBD, or PSP patients (for a complete update, visit www.molgen.ua.ac.be/FTDMutations).
4 Amyloid and Tau Deposition
4.1 Physical States of Aβ
In brain parenchyma, Aβ exist as monomers, dimers, low molecular weight oligomers, mycelia, protofibrils, and finally, mature densely compact fibrils (Fig. 1). While most of the Aβ assembly is extracellular, recent data also suggest that Aβ dimers could be constitutively secreted from neurons (93). Except the insoluble mature fibrils, all other precursor forms can be regarded as the soluble Aβ fraction, and most of these states including dimers but excluding monomers are toxic (94). While the soluble monomers and early aggregation states do not cross the threshold for immunohistochemical detection, densely packed aggregates are also less visible by immunohistochemistry due to epitope masking (see further). Thus, immunohistochemistry is most robust in identifying higher-order Aβ aggregation states including loosely arranged fibrils. Densely packed fibrils also acquire a β-pleated sheet conformation (95,96) and are therefore readily identified by silver impregnation techniques and Congo red or thioflavin (Th)-S or -T (95,96). Based on these different staining protocols, various types of extracellular and intracellular amyloid can be identified in both humans and mouse AD models.
4.2 Diffuse Deposits
Diffuse deposits are the first extracellularly visible Aβ deposits observed in AD and Down’s syndrome patients (11,80,97), and are also termed preamyloid deposits or preplaques (98). In AD brains, diffuse plaques are mainly present in the entorhinal cortex, allocortex (hippocampus and olfactory cortex), followed by the superficial layers of the neocortices, especially association neocortices (99). Occasionally, a cloud of diffuse plaques are observed under the cortical pia mater or the subependymal periventricular zones (99). These plaques also involve neostriatum and the molecular layer of cerebellar cortex (100). Various morphological forms of diffuse plaques have been described. However, in most frequent forms they appear as clusters of dots and rods with irregular borders merging diffusely with the parenchyma, and ranging in size from a few microns to many hundreds of microns (Fig. 2). Ultrastructural studies of such deposits have revealed scattered bundles of amyloid fibrils as well as amorphous, nonfibrillar material (101). Diffuse plaques frequently surround neuronal cell bodies and neurites without implicating vascular elements (101), suggesting that such deposits might be neuronally derived. Although it was shown that the progression to the end-stage of AD is associated with a proportional increase in fibrillar plaque isoforms (102), recent studies suggest independent evolution paths for fibrillar-dense and diffuse plaques (103). Biochemically, diffuse plaques seem to be composed of full-length and N-truncated Aβ42 (100,104). Moreover, diffuse plaques neither seem to associate with any cytoskeletal neuritic alterations nor display any significant astrogliosis or microglial activation (100,105,106). Only exceptionally, in end-stage diseases or in very aggressive familial forms of AD, such as Austrian T714I pathology, diffuse plaques are observed to be neuritic and associated with abundant glial and inflammatory pathology (76,100,107).
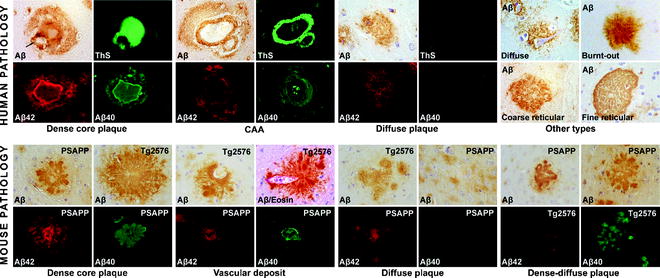
Fig. 2.
Amyloid pathology in AD patients and mouse models.
Upper panel: Spectrum of amyloid deposits in human AD pathology. Dense-core plaques typically have a ThS-positive compact core composed of Aβ40 and are typically surrounded by a corona composed chiefly of Aβ42. The center of the cores usually is faintly stained with anti-Aβ antibodies as a result of the densely packed fibrils. In the majority of the cases, a blood vessel can be identified on serial sections abutting to the dense core (arrow). CAA is ThS-positive, compact ring-like deposition of amyloid in the vessel wall that is chiefly composed of Aβ40 while the neighboring parenchymal diffuse amyloid is chiefly constituted of Aβ42. Diffuse-type of plaques are constituted of fine grains of nonfibrillar amyloid depositions, which is ThS-negative and mainly constituted of Aβ42. Normal appearing neurons can be sometimes discerned within diffuse plaques. Burnt-out types of plaques are represented by compact cores in a region devoid of neuronal bodies and neurites. Coarse reticular-type of plaques are composed of mesh-like dense-diffuse amyloid depositions, while a denser reticular pattern surrounded by a clear amyloid-parenchyma rim is described as fine reticular type of plaques.
Lower panel: Spectrum of amyloid deposits encountered in mouse models. Dense-core plaques usually consist of multiple smaller cores (PSAPP model) or more organized, single cores (APP model), but are distinct from human pathology as mice lack peri-core amyloid. The plaques are positive for Aβ40 and Aβ42, with the central dense regions staining faintly due to highly aggregated amyloid. Vascular amyloid deposition is a frequent finding in AD mouse models too, appearing as dysphoric angiopathy affecting small-to-medium sized blood vessels (shown here in Tg2576 mice) with Aβ40 predominating over Aβ42 (shown here in a PSAPP mouse). Diffuse plaques in mice biochemically and morphologically resemble human diffuse plaques being also ThS-negative and composed predominantly of Aβ42. Collections of diffuse plaques frequently occupy large areas in the parenchyma in PSAPP mice but more restricted in Tg2576. Dense-diffuse plaques are intermediate forms between diffuse and dense-core pathology with multiple nidi, not bridged as dense-core plaques, and sometimes surrounded by diffuse pathology (illustrated here in PSAPP mice). They are also chiefly composed of Aβ40 perhaps representing increasingly aggregating amyloid (in Tg2576 here).
AD mouse models based on APP and PS mutations also develop diffuse plaques that resemble human diffuse plaques biochemically and morphologically, in that they are also ThS-negative and predominantly composed of Aβ42. In mice, diffuse deposits can exist as small patches of amyloid, but given the supraphysiological levels of Aβ production, the number of plaques increases rather rapidly to occupy large areas of brain parenchyma. All brain regions can be affected, although the first region to be involved largely depends on the specific promoter utilized. Certain brain regions such as cerebellum and basal ganglia seem to have a paucity of diffuse deposits (further discussed under section Mouse Models).
4.3 Compact Amyloid Deposits with Dense Cores
Dense-cored plaques were first observed in 1892 and named “senile plaques” in 1911 by Simchowicz (108). These plaques consist of a dense core of amyloid fibrils arranged radially, surrounded by an immediate clear halo and an external belt of amyloid. Sometimes, long arms of amyloid connect the surrounding corona with the inner core of the plaque. The central core regions are one of the most compact forms of amyloid observed in human brain, and are readily stained with Congo-red or ThS, appearing round, star, or cross-shaped (“Malthese cross”). With Aβ immunohistochemistry, the dense-core regions stain faintly perhaps due to the masking of Aβ epitopes. This type of dense aggregated cored plaques is the most characteristic finding in the Flemish APP pathology, with the “largest” dense-cored plaques encountered in AD (41). Dense-core plaques are also called neuritic plaques as they are frequently surrounded by dystrophic neurites with or without NFT pathology or neuropil threads and also seem to incite both astrocytosis and microgliosis (99). Occasionally, dense-core plaques seem to lack a coronal plaques region as well as dystrophic neurites and these compact cores are sometimes referred to as “burnt-out” or “end-stage” plaques with the belief that the surrounding neuronal elements are destroyed in these plaques. Biochemically, while the corona of the plaques has predominantly, if not solely, Aβ42, the central dense-core regions are rich in Aβ40. Thus, Aβ42 is present in both the core and the corona of senile plaques although in different proportions, but it seems that increased Aβ42 is a common feature for both normal aging and senile AD cored-type of deposits (109). It is possible that with increasing compactness and aggregation state of the plaque cores, an increasing ratio of Aβ40/Aβ42 occurs for the deposited amyloid.
The highest number of dense-cored plaques is found in the superior hippocampal regions (CA1–CA2), subiculum, layers II, III, and V of the entorhinal cortex, and the plaques become fewer and larger towards CA4 (110,111). Dense plaques together with reticular plaques (see later) are also observed in lower cortical layers of the association cortices, such as the temporal association cortex. Primary motor and visual cortices also deposit mostly dense-core plaques. In the basal ganglia, pallidum is usually the only striatal nucleus where dense plaques appear. In cerebellum, senile plaques are abundant in the Purkinje and granular cell layers (100) and, interestingly, the highest incidence of amyloid plaques in the cerebellum is found in presenile patients (112). Substantia nigra and other brainstem nuclei also deposit dense plaques and the deposition is proportional to the amyloid burden found in cerebellum (112).
Dense-cored plaques are also observed in mouse models, especially in the environment of a high Aβ40/Aβ42 ratio (81). Importantly, dense-cored plaques lack the characteristic surrounded corona of diffuse amyloid, and frequently show presence of multiple small cores, especially in PSAPP mice where Aβ42 levels are high. Biochemically, the dense cores resemble human dense senile cores being ThS positive and rich in Aβ40. As mentioned, peri-plaque corona is mostly absent, but when present, is predominantly of Aβ42-type as shown in AD. Interestingly, dense-core plaques in mouse models most resemble similar plaques observed in the Flemish APP pathology (34). Dense-cored plaques are observed in all regions of the mouse brain with a preference for the hippocampal region and the hippocampal sulcus (34,107).
4.4 Compact Deposits Without Amyloid Cores
Compact deposits lacking a discernable amyloid core are also called “primitive plaques” due to an arguable viewpoint that they progress in time to form dense-core plaques. Such plaques are the most common type of plaques in the cerebral cortex in most AD patients and consist of agglomerated wisps of amyloid deposits arranged in a reticular pattern (99). Typically, these plaques contain almost exclusively Aβ42. When the faint reticular pattern has no readily demarcated edge, these are called “coarse” reticular plaques, and seem to predominate in AD, especially in FAD patients such as those harboring PS1 G384A mutation (113). When the reticular pattern is clearly demarcated by a strong amyloid edge on immunohistochemistry, these are called “fine” reticular dense plaque. The latter type of plaques is the most frequent compact, noncored plaque found in the Flemish APP pathology (41) (see further).
In mice models, diffuse plaques are seen especially in very early ages, and dense-diffuse plaques are more common (34). The compacted region contains more Aβ40, and diffuse deposits are predominantly Aβ42-positive. Dense-diffuse deposits can appear throughout the mouse brain, with a relative sparing of the basal nuclei and cerebellum.
4.5 Vascular Amyloid Deposition
When Aβ is deposited in the walls of small-to-medium-sized blood vessels of the brain and of leptomeninges and incites a pathological response, it is referred to as cerebral (congophilic or ThS-positive) amyloid angiopathy of the Aβ type (114). More than 10% of the persons over 60 years of age and ≈80% of the AD patients have at least some degree of CAA (115). CAA affects most frequently the occipital, temporal, and frontal lobes, being less frequently found in hippocampus, cerebellum, and basal ganglia, and almost absent in the white matter and brain stem (116). Leptomeningeal arteries are most frequently involved, followed by cortical arteries, arterioles, and capillaries, while the venous sector is less frequently involved (116). While Aβ deposits seem to appear in the media near the smooth muscle cells in larger vessels, capillary CAA (cap-CAA) has Aβ deposited first on the abluminal side of the basement membrane (117). Interestingly, cap-CAA have frequently small perivascular caps of amyloid or perivascular amyloid fibrils radiating in the surrounding brain tissue, described as dysphoric angiopathy or “drusige Entartung” (114,118,119). In pathologies where CAA is a prevailing feature such as Flemish APP mutation, dysphoric angiopathy is also observed within larger vessels (41). Biochemically, Aβ40 is the major constituent of CAA affecting large leptomeningeal and neocortical vessels, while both Aβ40 and Aβ42 are present in CAA affecting smaller parenchymal vessels.
Vascular Aβ depositions also cause secondary changes in vessels such as hyalinization and degeneration as indicated by increased representation of collagen-IV in the basement membrane. Not infrequently, “double-barreling” or “vessel-within-vessel” configuration is also observed due to deposition of Aβ both in the vessel wall and under glia limitans, separated by the distended perivascular spaces (116,120). Thus, not surprisingly, CAA accounts for approximately 10% of all intracerebral nonfatal hemorrhages in the elderly (121). Interestingly, CAA also seems to be capable of initiating a strong local neuritic and inflammatory pathology (86) and in this respect cap-CAA, but not CAA associated with larger vessels, seems to correlate well with parenchymal pathology (119,122).
4.6 Intraneuronal Aβ
Recent data also indicate that Aβ is deposited within neurons in brains of AD and Down’s syndrome patients (126,127). While this is a relatively new and understudied area of amyloidosis, it is highly likely that intraneuronal Aβ, similar to a plethora of inclusion proteinopathy diseases, may induce cytopathic effects with important roles in AD pathogenesis. Several mouse models have been established that deposit intraneuronal Aβ either solely or in early stages of diseases and shall be discussed in relevant sections.
4.7 NFT and NF Threads
Tau is the major intraneuronal proteopathy in AD inside the body of the neurons (as NFTs) or along dystrophic neurites (as neurophil threads). Early studies on tau deposits recognized phosphorylated tau as the major component of PHF (128–131). In AD, PHF consist of 10–20 nm filaments distributed especially in the entorhinal cortex, hippocampus, amygdala, and association temporal cortex (100). Staging of AD based on the prevalence of NFT pathology is also proposed, which seems to correlate better with the cognitive deficits compared to criterions based on amyloid deposits (132,133).
Tau is also the main pathological finding in FTLD-tau disorders such as Pick’s disease. The histopathological features of Pick’s disease are dominated by argyrophilic tau-positive inclusions (Pick bodies) and swollen achromatic cells (Pick cells). They usually occur in the granular layer of dentate gyrus, pyramidal cells of the CA1, subicular hippocampal sectors, neocortex, and in several subcortical nuclei. In the neocortex, they predominate in the layers II and VI, in contrast to the neurofibrillary tangles in AD that appear especially in layers III and V. By electron microscopy, Pick bodies contain intermediate filaments, 15 nm straight filaments, and some paired helical filaments (134). Biochemical characterization showed that the insoluble tau in Pick bodies consists of 3R-tau isoforms (135,136). However, recent studies have demonstrated much greater biochemical heterogeneity and up to 50% of patients with Pick’s disease having either at least as much 4R as 3R-tau, or even a predominance of 4R-tau (49,137,138). Mouse models expressing various isoforms of tau are discussed hereunder.
5 Mouse Models of Dementia
Several mouse models of AD have been established based on FAD mutations that develop amyloid pathology. The discovery of tau mutations in FTDP-17 families also brought new perspectives with regard to common tau dysfunctions in AD and FTDP-17 (88). We have reviewed below some of the most important mouse models based on these pathogenic mutations that show how these models have expanded our molecular understanding of diseases like AD or FTD. For a complete list of AD mouse models, visit www.alzforum.org/res/com/tra.
5.1 APP Mouse Models
5.1.1 PDAPP and APP/Ld2 (APPIndiana and APPLondon) Models
The PDAPP mouse model was the first successful mouse model established in the study of AD. It carries the human APP770 with the Indiana V717F mutation under the control of human platelet-derived growth factor-β (PDGF-β) neuronal promoter and on a mixed C57BL/6, DBA, and Swiss-Webster genetic background (123). More than tenfold overexpression of the APP transgene occurred compared to the murine endogenous APP levels. The high expression level is perhaps due to the transgenic construct utilizing a splicing cassette that permits the expression of all three major APP isoforms. Also, the transgene is predominantly expressed in neurons in the cortex, hippocampus, hypothalamus, and cerebellum. Amyloid pathology is detected around 6–9 months of age, beginning in the hippocampus, corpus callosum, and cerebral cortex. The deposits range from diffuse to ThS-positive dense plaques and CAA, and the size and densities of these deposits increase with age. Dystrophic neurites are present in the vicinity of amyloid depositions; however, no NFT is identified. GFAP-positive activated astrocytes surround the plaques while isolated, activated microglia are detected throughout the neocortex. Synaptic and dendritic densities are reduced in the molecular layer of the dentate gyrus, but no significant neuronal loss is recorded here or in the neocortex. Behavioral deficits begin to appear around 3 months of age, with impairments in visuospatial reference memory (139), spatial discrimination tasks, spontaneous object-recognition, and operant learning (140). As we shall see later for other mouse models as well, behavioral deficits occur before immunohistochemically detectable amyloid depositions appear in brain (Table 1).
Table 1
Neuropathology of commonly utilized mouse models in Alzheimer’s disease and other tauopathies
Protein model | Line/Reference | Genetic background | Promoter | Associated pathology | Cognitive and behavioral changes |
|---|---|---|---|---|---|
APP models | PDAPP (124) | Human APP770 with Indiana (V717F) mutation | PDGF-β | Amyloid depositions at 6–9 months; mostly diffuse deposits (Aβ42 rich) and CAA (Aβ40 rich). Significant loss of SMCs and hemorrhages; no neuronal loss; no NFT | Impairments in visuospatial reference memory, spatial discrimination tasks, spontaneous object-recognition and operant learning at ≈3 months |
APP/Ld2 (142) | Human APP695 with London (V717I) mutation | Thy-1 | Amyloid plaques and CAA at 10–12 months. Loss of SMCs but no hemorrhages; cholinergic fiber distortions | Alternating episodes of hyperactivity, anxiety, aggression, decreased exploration, and ambulation starting at ≈8 weeks | |
Tg2576 (125) | Human APP695 with Swedish double (K670N/M671L) mutation | Hamster PrP | Plaque at ≈9–12 months; mostly dense deposits and CAA (Aβ40 rich). Loss of SMCs and hemorrhages; no neuronal loss; no NFT | Spatial alternation task and longer escape latencies in the water maze at ≈9–10 months | |
APP23 (126) | Human APP751 with Swedish mutation | Thy-1 | Amyloid deposition at 6 months; mostly CAA and dense plaques. Loss of SMCs and hemorrhages; neurodegenerative changes with hyperphosphorylated tau, but no NFT | Severely decreased learning and training performances at 3 months | |
Austrian (149) | Human APP695 with Austrian (T714I) mutation | PDGF-β | Intraneuronal Aβ in hippocampus by 6 months. Intraneuronal N-truncated Aβ42 co-localize with endosomal-lysosomal pathway markers; brain volume reduction on MRI | Subtle behavioral impairments in light–dark transition box tests at 12 months | |
TgCRND8 (154) | Human APP695 with Swedish and Indiana mutations | Hamster PrP | Amyloid depositions at 6 weeks in subiculum and frontal cortex; dense cored deposits appear first in subiculum, while diffuse depositions appear later. Neuritic pathology and synaptic degeneration present | Impairment in the acquisition of spatial information during place discrimination training at≈11 months | |
APPDutch (156) | Human APP with Dutch (E693Q) mutation | Thy-1 | Amyloid pathology around 22–25 months; most depositions as CAA where mutant DutchAβ40 predominates. Loss of SMCs and hemorrhages | NA | |
Tg-SwDI (157) | Human APP770 with Swedish, Dutch and Iowa (D694N) mutations | Thy-1 | Amyloid pathology starts at 3 months with diffuse-like plaques and vascular accumulations in thalamic region at 6 months; Decreased Aβ42/Aβ40 ratio | NA | |
hAPP-Sw/Ind (159) | Human APP with Swedish and Indiana mutations | PDGF-β | Amyloid pathology at 7 months with dense-like deposits and vascular deposition. Dystrophic neurites present | NA | |
hAPP-Arc/Sw/Ind (159) | Human APP with Arctic (E693G), Swedish, and Indiana mutations | PDGF-β | Amyloid deposition starts around2.3 months and is associated with dystrophic neurites. Introducing Arctic mutation in Sw/Ind mice decreases the Aβ42/Aβ40 ratio, and favors parenchymal depositions | NA | |
PSAPP models | PS1 (159) | Human PS1 with M146L or M146V mutation | PDGF-β | No amyloid pathology up to 12 months; Higher Aβ42/Aβ40 ratio | No behavioral abnormalities up to 12 months |
Tg2576 × PS (162) | Human APP695 with Swedish mutation Human PS1 with M156L mutation | Hamster PrP | Accelerated amyloid pathology with smaller and more numerous dense plaques, less diffuse depositions. Increased Aβ42/Aβ40 ratio | Impaired Y-maze spontaneous alternations at ≈3 months | |
APPSw × PS (161) | Human APP with Swedish mutation and human PS1 with A246E mutation | Mouse PrP | Accelerated amyloid pathology with smaller and more numerous dense plaques, less diffuse depositions. Increased Aβ42/Aβ40 ratio | NA | |
APP/Ld × PS1(164) | Human APP695 with London mutation and human PS1 with A246E mutation | Thy-1 | Dense amyloid cores and CAA load increased more than double compared to APP/Ld mice. Increased soluble brain Aβ levels | NA | |
TgCRND8 × PS (154) | Human APP with Swedish and Indiana mutations and human PS1 with M146L and L286V mutations | Hamster PrP | Accelerated amyloid plaque pathology appearing at 4–6 weeks | NA | |
APPDutch × PS1 (156) | Human APP with Dutch mutation and human PS1 with G384A mutation | Thy-1 | Amyloid pathology around 6 months as both diffuse and compact plaques (DutchAβ42 twice as abundant as DutchAβ40) | NA | |
APP × PS1-knockin (152) | PS1 M233T and L235P knock-in and human APP751 with London and Swedish mutations | Thy-1 (PS1 knockin) | First Aβ depositions at 2.5 months that become generalized by 6 months. Amyloid deposits associated gliosis and distrophic neurites; Severe neuronal loss in hippocampus by 10 months | NA | |
Aβ-only expressing mice | Bri-Aβ42/Bri-Aβ40 (166) | Fusion BRI and Aβ42 (or Aβ42) protein | Mouse PrP | Bri-Aβ42 mice start depositing compact plaques and CAA at 3 months in the cerebellum and at 6 months in the cortex. Diffuse pathology appears with increasing age. Dense plaques associated with reactive astrogliosis and rare dystrophic neurites | NA |
Tau models | JNPL3 (167) | Human four repeat tau with P301L mutation | Mouse PrP | NFT in motor neurons in the spinal cord at ≈6.5 months and progress to neuronal loss in brain stem and midbrain | Absence of escape extension during tail elevation at ≈5 months, weakness in all limbs, dystonic postures, decreased vocalization, and decreased weight |
JNPL3 × Tg2576 (170) | Human four repeat tau with P301L mutation and human APP with Swedish mutation | Mouse PrP | Amyloid depositions comparable with those of Tg2576 mice. Significant increase of NFT as well as insoluble tau levels in limbic regions compared to JNPL3 mice | Same as for the JNPL3 × Tg2576 mice | |
APP Sw/Tau P301L/PS1M146V (153) | Human four repeat tau with P30lL mutation in knock-in PS1 (M146V) | Thy-1 (PS1 knockin) | Intraneuronal Aβ deposit at 3 months and extracellular plaques at 6 months. At 18 months NFT occur in the hippocampus | Retention/retrieval deficits appear at ≈4 months |
Four years later, a model carrying another mutation on the same codon, the V717I London APP mutation, was published as the APP/Ld2 model (141). This transgenic mouse strain carried the V642I mutation on the APP695 isoform under the regulation of the mouse thymus cell antigen 1 promoter (Thy-1) and on a FVB/N background. Human APP insert was expressed two- to fivefold higher than the APP endogenous level in hippocampus and neocortex. However, in contrast to the PDGF promoter (utilized in the PDAPP model) that drives expression predominantly in the neocortex, the murine Thy-1 promoter drives transgene expression more widely, including expression in the subcortical regions. Amyloid deposition occurred around 12 months of age as both diffuse and compact neuritic plaques were most abundant in the hippocampus and cortex, and only occasionally in thalamus and white matter. APP/Ld2 mice also develop significant vascular amyloid depositions primarily in pial, cortical, thalamic, and hippocampal vessels with predominant Aβ40 accumulation (142). Behavioral deficits begin to appear around the age of 8 weeks with alternating episodes of hyperactivity, anxiety, aggression, decreased exploration, and ambulation (141).
5.1.2 Tg2576 and APP23 (APPSwedish) Models
The Tg2576 mouse model expresses human APP695 harboring the Swedish double mutation (K670N/M671L) under the control of the hamster prion protein (PrP) promoter and on a C57B6/SJL background (124). Tg2576 expresses transgenic APP sixfold over the endogenous murine APP level. Total Aβ secretion increases three- to eightfold, and although cellular secretion of Aβ42/Aβ40 ratio is not changed, these mice predominantly deposit Aβ40 (143). Amyloid pathology is not detected in animals below 3 months of age. Around 9–10 months, amyloid deposits can be identified beginning in the entorhinal and piriform cortices and continuing in the neocortex and cerebellum, in a pattern similar to that of the PDAPP model. These consist mainly of large dense-core plaques and CAA (rich in Aβ40), and occasionally of diffuse-like deposits (rich in Aβ42). Regions that deposit most of the dense deposits were neocortex and subiculum. In the thalamic region, there is an abundance of CAA and cap-CAA type of depositions (34). Compared to the PDAPP model that carries the London APP mutation and thus causes decreased Aβ40 production, this model increases the absolute amounts of Aβ40 and corroborates with a more compact amyloid pathology. Amyloid depositions are surrounded by dystrophic neurites and gliotic changes. Neither NFTs nor neuronal loss could be identified (144). Nevertheless, memory deficits such as poor performance in spatial alternation task and longer escape latencies to the hidden platform in the water maze are observed at 9–10 months (124).
Like the Tg2576 mouse model, the APP23 model also carries the Swedish double mutation, but in the APP751 human isoforms and under the control of murine Thy-1 promoter, and on a C57BL/6 background (30,125). In these mice, the transgene expression is sevenfold over murine APP levels and leads to amyloid deposits beginning with ≈6 months of age in the hippocampus, and later involving the neocortex, thalamus, and olfactory bulbs. APP23 mice also present with alterations of the cholinergic system, synaptic button loss, and neuronal loss (145). As mice age, they also deposit vascular amyloid in the majority of the pial vessels and in a significant fraction of the vessels in thalamus, cortex, and hippocampus (30). In these mice, vascular amyloid is associated with SMC degeneration leading to microhemorrhages, and they have been extensively utilized as a model for CAA with spontaneous hemorrhagic stroke (33,146). APP23 mice also show severely decreased learning and training performances by 3 months of age (147).
5.1.3 APP/Austrian and Similar Models
The APP/Austrian mouse model (APP-Au) (148) was based on identification of the APP (T714I) mutation in an Austrian family, with one of the earliest age of onsets in AD of ≈34 years, the patients having abundant intracellular and extracellular amyloid deposits in the brain (76). The latter, strikingly, was nonfibrillar diffuse amyloid, composed of N-truncated Aβ42 in the absence of Aβ40, as shown in other mouse models (149). In vitro, this mutation led to one of the highest Aβ42/Aβ40 ratios among all familial AD mutations studied (76,77). The APP-Au mouse model carries the T714I mutation on a human APP695 isoform, under the control of the PDGF-β promoter. Despite having tenfold lower transgene expression than endogenous murine APP, intraneuronal hippocampal Aβ deposits appear by 6 months of age. Accumulations increase with age, and parallel decreased brain sizes on volumetric MRI, compared to age-matched and similar transgene-expressing APP wild-type mice. Subtle behavioral impairments were noted in the light–dark transition box tests. In this model, the majority of the intraneuronal Aβ deposits colocalized with markers of endosomal–lysosomal pathway, supporting the hypothesis that intraneuronal accumulation of Aβ could be an important factor in AD pathogenesis. Interestingly, mice expressing both Austrian and Swedish double mutations despite having fivefold higher transgene levels comparted to APP/Au mice have significantly lower intraneuronal Aβ and lack the reduced brain volume phenotype (150). APP/Sw is processed in the secretory pathway instead of the endosomal–lysosomal pathway necessary for intraneuronal Aβ accumulation thereby suggesting that intraneuronal Aβ accumulation is related to reduced brain volumes in APP-Au mice. These data also find support in mouse models based on mutant PS1-knockin where intraneuronal N-truncated Aβ42 (but not extracellular Aβ40 or Aβ42) could correlate with neuronal loss (151,152) (see later).
5.1.4 TgCRND8 (APPSwedish/Indiana) Model
The TgCRND8 line carries the Swedish double mutation and the Indiana (V717F) mutation on a human APP695 isoform under the control of the hamster PrP promoter, and on a C3H/B6 background (153). γ-Secretase site APP mutations lead to a drastic reduction of Aβ40 or total Aβ (76,77), and Swedish APP mutation ensures a high Aβ secretion level, for instance, levels of Aβ42 in 6-month-old TgCRND8 mice are close to those seen in PDAPP mice at 16 months of age. Amyloid deposition started at ≈6 weeks of age in TgCRND8 mice, appearing first in the subiculum and frontal cortex, and later also involving corpus callosum, hippocampus, dentate gyrus, olfactory bulbs, and pial vessels. Thalamus, striatum, and cerebellum were the last regions involved where amyloid deposited at 16–35 weeks of age. The first amyloid deposits appeared as cored deposits in subiculum, changing to larger, multicored deposits with age. Diffuse pathology occurred in the cortex around 35 weeks, and by 45 weeks, diffuse amyloid depositions appeared throughout the cortex. In the olfactory bulbs, diffuse pathology occurred even earlier but remained unchanged until late ages. Dense plaques associated with dystrophic neurites, synaptic degeneration, and inflammatory response. From 11 weeks onwards, TgCRND8 mice show impairment in the acquisition of spatial information during place discrimination training (153).
5.1.5 APP/Dutch Model
The E693Q Dutch APP mutation leads to extensive CAA with recurrent cerebral hemorrhages and dementia in the HCHWA-D syndrome. Although earlier attempts with Dutch APP transgenesis did not show amyloid deposition till 18 months of age (154), a new transgenic mouse model based on this mutation has recently been generated that closely mimics human HCHWA-D pathology (155). The Dutch mice drive APP751 with the E693Q mutation under the control of murine Thy-1 promoter, and with a fivefold transgenic overexpression compared to murine APP levels. The transgenic expression, restricted to neurons and neuronal processes, was predominantly observed in the neocortex, hippocampus, and brain stem. Dutch Aβ40 was readily detected in 23-months-old mice, and vascular amyloid depositions could be also identified around this age. CAA appeared first in the leptomeningeal vessels followed by cortical vessels, while parenchymal depositions were restricted to a few diffuse accumulations. Affected vessels showed a severe loss of SMC with multiple surrounding fresh hemorrhages, and were associated with a strong perivascular microglial inflammatory reaction.
5.1.6 Tg-SwDI (APPSwedish/Dutch/Iowa) and Similar Lines
A step forward in studying the relation between Aβ and the vascular compartment was the introduction of two vasotropic APP mutations in a single construct. The Tg-SwDI model harbors human APP770 carrying the Swedish APP mutation together with the Dutch and Iowa (D694N) mutations, under the control of the Thy-1 murine promoter and on a C57Bl/6 background (156). The transgene expression was less than 50% of the level of endogenous mouse APP. Starting with 3 months of age onwards, mice had numerous amyloid deposits appearing first in subiculum, hippocampus, and cortex, and by 6 months of age also involved olfactory bulbs and thalamus. CAA involving predominantly cerebral microvessels but also large meningeal vessels appeared at about 6 months and was associated with reduced microvessel densities, endothelial loss, accumulation of inflammatory cells, and occasionally, microhemorrhages (157). Mutant Aβ40 predominated and although parenchymal accumulations presented as diffuse-like depositions, interestingly, all ThS-positive amyloid deposits colocalized with blood vessels (157).
Another recent mouse line expressing vasotropic human Aβ was APP Arctic (E693G) mutation along with APP Swedish and APP Indiana mutations, Tg-SwAI, under the control of the PDGF-β promoter and on a C57BL/6J background (158). This line had decreased Aβ42/Aβ40 ratios and induced neuritic amyloid plaques at 2.3 months, which was earlier and more extensive than in the Tg-SwDI line. In contrast to APP Dutch mice that deposits almost exclusively vascular amyloid with a predominance of Aβ40 in the parenchyma, Tg-SwAI mice also have high Aβ40 levels but develop only parenchymal plaques with no obvious increase in the CAA formation, a phenomenon that seems to indicate that fibrillogenetic properties of a specific mutant Aβ species may be a prime factor in its preferential vascular vs. parenchymal deposition.
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


