and Luc Buée1, 2, 3
(1)
Univ Lille Nord de France, Lille, France
(2)
Inserm-U837, Lille, France
(3)
UDSL, Faculté de Médecine, Institut de Médecine prédictive et de Recherche thérapeutique, Centre de Recherches Jean-Pierre Aubert, Lille, France
Abstract
Tau pathology refers to molecular mechanisms leading to the intracellular aggregation of abnormally modified Tau protein isoforms and to the propagation of this degenerating process along neuronal circuitry. Tau proteins belong to the family of microtubule-associated proteins. Tau is strongly expressed in neurons, localized in the axon and is essential for neuronal architecture, plasticity and network. In animal models, overexpression of mutated Tau proteins is often used to induce a Tau pathology, leading to motor and/or cognitive dysfunction. These animal models are essential to understand how neuronal degeneration and Tau aggregation are related. Indeed, Tau pathology is certainly a good therapeutic target, but untangling Tau remains a major therapeutic challenge.
Key words
Alzheimer’s diseaseNeurofibrillary degenerationMicrotubule-associated protein TauTauopathiesTau pathology1 Introduction
In Alzheimer’s disease (AD), microtubule-associated protein Tau is the major component of paired helical filaments inside degenerating neurons referred to as neurofibrillary tangles (NFT). However, aggregation of Tau inside neurons or glial cells is observed in more than 20 neurological disorders, which together form the so-called Tauopathies. The Tau/MAPT gene locus is associated with the development of frontotemporal dementia, and microdeletion in the locus region is associated with mental retardation. Besides its most well-known function as being a microtubule-polymerizing and -stabilizing factor, Tau is implicated in the architecture and organization of the paralleled order of microtubules and it also important for the regulation axonal transport through the regulation of motor proteins and vesicular transport. When aggregated, one of the major questions that remain to be answered is whether there is a loss or gain of toxic functions. Recent development of transgenic animal models may be useful to address this question.
2 Microtubule-Associated Tau
2.1 Tau/MAPT Gene
The human Tau/MAPT gene is unique, and contains 16 exons located over more than 150 kb on the long arm of chromosome 17 at band position 17q21 (1, 2) (Fig. 1). Although the mechanism underlying the neuronal expression of Tau is not known, the expression of Cre recombinase under the control of the murine Tau promoter results in an almost completely restricted expression of the recombinase in neurons (3). In humans, Tau expression is also found in muscle and scarcely in other tissues or organs (4). Several polymorphisms are identified and found to be in complete linkage disequilibrium with one another, defining an extended haplotype that covers the entire MAPT gene (5) and even spans to a region covering ∼1.8 Mb (6) (Fig. 1a). The H2 haplotype is rarer than the H1 haplotype (73–79%) in healthy individuals, and results from H1 by the inversion of a ∼970 kb segment (7). The H2 haplotype is associated with an increased risk of de novo microdeletion and developmental delay and learning disability (8, 9). Specific H1 sub-haplotypes are associated with the risk of developing AD, Parkinsonism syndromes such as corticobasal degeneration and progressive supranuclear palsy and likely influence the profile of Tau isoform expression (10–12).
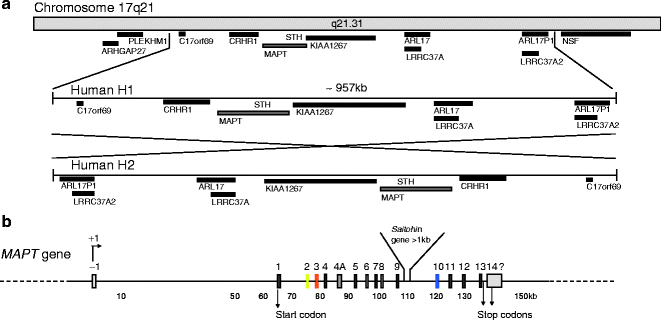
Fig. 1.
Chromosome localization and microtubule-associated MAPT/Tau gene
(a) Chromosome localization and haplotypes of Tau. Tau gene is located on chromosome 17 at position q21.31. The locus of the human Tau gene occurs as two haplotypes H1 and the inverted version of H1, H2. No recombination is observed between H1 and H2 over a region of approximately 1.5 Mb. Tau gene locus and the surrounding genes are represented. H1 and its ∼970 kb inverted H2 haplotype are represented. (b) Tau gene. Tau gene spans more than 130 kb and is composed of 16 exons. Transcript initiation starts at exon -1 and the start codon is located in exon 1. They are two alternate stop codons located following exon 13 or inside exon 14. The 3′ ending of exon 14 is not completely characterized in humans. The initiation of transcription is indicated by +1. The promoter region of Tau encompasses the sequence upstream exon -1, the exon -1, and the intron 0. The promoter is GC-rich and contains SP1 and AP2 binding-motifs proximal to exon -1.
2.2 Posttranscriptional Modifications
The Tau primary transcript contains 16 exons (Fig. 2a). In the central nervous system (CNS), two transcripts of 2 kb and 6 kb arise from the use of two alternative polyadenylation sites, the 2 kb mRNA targets Tau to the nucleus and the 6 kb encodes the major form in axons (13, 14). In the human brain, Tau exons 4A and 8 are skipped. Exon 4A is found in bovine, human, and rodent peripheral tissues with a high degree of homology. Exon 8 is alternative and found in striated muscle, spinal cord and pituitary (4). Noteworthy, Tau expression is tenfold higher in brain and spinal cord than in skeletal muscle. Cryptic splicing sites are described in exon 6 that generate Tau mRNA lacking the remaining 3′ exon cassettes (15). Those are found in muscle and the spinal cord, but the presence of the protein remains to be determined. Exon –1 and adjacent intron are part of the promoter. Exon –1 is transcribed but not translated. Exons 1, 4, 5, 7, 9, 11, 12, and 13 are constitutive exons. Exon 14 is part of the 3′ untranslated region of Tau mRNA (2, 16, 17). Exons 2, 3, and 10 are alternatively spliced and are adult brain-specific (18). Exon 3 is never included independently of exon 2, although exon 2 can be included independently of exon 3 (2). Thus, alternative splicing of these three major exons allows for six mRNAs (2–3–10-; 2+3–10–; 2+3+10–; 2–3–10+; 2+3–10+; 2+3+10+) and in the human brain, the Tau primary transcript comprises six mRNAs translated into six protein isoforms (16, 17, 19) (Fig. 2b).
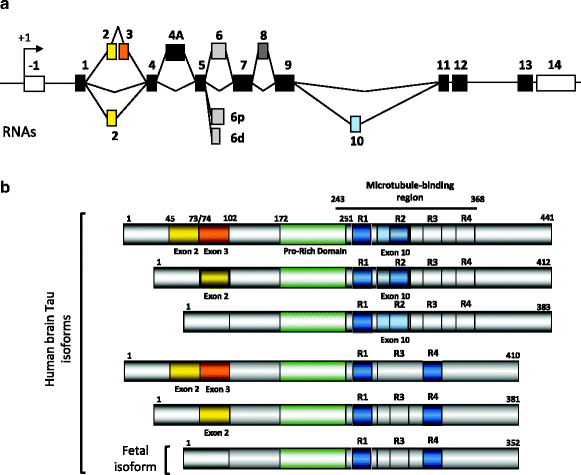
Fig. 2.
Alternative splicing and Tau proteins in the central nervous system
(a) Alternatively spliced cassettes. Several mRNAs are generated by alternative splicing of exons 2, 3, 4A and 10. The 6p and 6d are cryptic splicing sites rarely used. The selection of those sites is supposed to generate carboxy-truncated Tau proteins (for review see (13)). Exon 4A is excluded in the brain and included in the spinal cord and peripheral nervous system. Exon 8 is found included only in muscle and found also in other species such as bovine Tau. (b) Tau protein isoforms in the central nervous system. In the human brain, six major Tau isoforms are generated from the alterative splicing of exons 2, 3 and 10. Exon 3 is always included with exon 2. The exon 10 encodes an additional microtubule-binding motif numbered R1 to R4 that together defines the microtubule-binding region. Half of Tau proteins contain three microtubule-binding motifs (light grey) and the other half (dark grey) has four microtubule-binding motifs. Upstream the microtubule-binding is the proline-rich domain containing several phosphorylation sites that regulate Tau binding to microtubules.
3 Tau Proteins, Posttranslational Modification, and Functions
3.1 Tau Functions
Tau was first discovered in the mid-1970s as a factor promoting tubulin polymerization (20). Tau is a neuronal protein essentially located within the axonal compartment. Its structure makes it essential for the organization, stabilization and dynamics of microtubules. The primary sequence of Tau can be subdivided in an amino-terminal region followed by a proline-rich domain, the microtubule-binding repeat motifs and the carboxy-terminal tail (Fig. 2b). Regarding the primary structure, the polypeptide sequences encoded by exons 2/3 add to Tau acidity, whereas exon 10 encodes a positively charged sequence that adds to the basic character of Tau. More generally, the amino-terminal region has a pI of 3.8, followed by the proline domain, which has a pI of 11.4, and the carboxy-terminal region is also positively charged with a pI of 10.8. Tau is rather a dipole with two domains with opposite charge, which can be modulated by posttranslational modifications. Structural analysis of human Tau using several biophysical methods showed that in solution Tau behaves as an unfolded protein (21). However, functions of Tau are distributed both in the amino- and the repeat-domains.
The amino-terminal region together with the proline-rich domain is referred to as the “projection domain.” This unstructured and negatively charged region detaches from the surface of microtubules (22) and can interact with the plasma membrane or cytoskeletal proteins (23). Tau may also contribute to the spacing in-between microtubule lattice and also to the parallel ordered organization of microtubules in axons (24). The spacing may be dependent upon the presence of additional amino-terminal sequences such as exons 2, 3 or 4A. The latter is included only in the spinal cord in peripheral nerve tissue (25). More recently, using a biophysical assay, Rosenberg and colleagues suggest that the amino-terminal regions of two Tau molecules, each one individually binding to a microtubule, form an electrostatic “zipper” (26). The amino-terminal region of Tau also interacts with a growing panel of proteins including motor proteins such as kinesin-1 (27) and dynactin–dynein complex (28), SH3-containing tyrosine kinases such as the phospholipase C-gamma 1 or the p85α subunit of PI-3K (29). Recently, it has been established that Tau regulates the motility of dynein and kinesin motor proteins by an isoform-dependent mechanism. Indeed, the shortest Tau isoform lacking exons 2, 3 and 10 impedes the motility of both kinesin and dynein, whereas the longest Tau isoforms with all exons less affect motor protein motility (30). Thus, the axonal transport of vesicles may be finely tuned by the ratio of Tau isoforms expressed. In neurodegenerative disorders, a modified pattern of Tau isoform expression/ratio, due to Tau aggregation for instance, may profoundly affect the axonal transport and could possibly lead to neurodegeneration (31).
The carboxy-terminal region, which is the basic region of Tau protein, is characterized by the presence of three or four repeat motifs, depending on the inclusion or not of the exon 10 encoding sequence. The repeat motifs are corresponding to the microtubule-binding domains. Apart from binding to microtubules, the repeat-domains of Tau interact with the histone deacetylase 6 (HDAC6), and HDAC6 is suggested to regulate Tau phosphorylation. The inhibition of the proteasome activity by MG132 enhances the interaction of HDAC6 to Tau. Interestingly, in this condition, HDAC6/Tau complexes are observed in perinuclear aggresomes (32). The proteasomal degradation of Tau is also sensitive to chaperone and co-chaperones Hsc70 and BAG-1 (Bcl2-associated athanogene-1) respectively that are interactors of Tau proteins (33, 34). The repeat-domains also interact more strongly with apolipoprotein E3 (ApoE3) than the E4 (ApoE4) isovariant (35). The functionality of Tau–ApoE interaction remains unknown. However, a triple transgenic mouse model of AD crossed with a knock-in ApoE4 mouse showed a strong influence of ApoE4 upon the topographical distribution of amyloid deposits, but surprisingly, the Tau pathology was strongly reduced (36). Together, Tau interacting partners may profoundly regulate its function but also modulate the Tau pathology. The physiologic and pathologic functions of Tau are also regulated by posttranslational modifications such as phosphorylation.
3.2 Tau Phosphorylation
Microtubule-associated proteins Tau are phosphoproteins (37). There are 85 potential phosphorylation sites on the longest Tau isoform. Phosphorylation sites were characterized using phospho-dependant Tau antibodies, phospho-peptide mapping, mass spectrometry, or nuclear magnetic resonance (NMR). According to the latest extensive analysis of Tau phosphorylation (38) and that of previously published reviews (39, 40), 71 among the 85 putative phosphorylation sites can be phosphorylated in physiological or pathological conditions (for review see (41)). Most of the phosphorylation sites are surrounding the microtubule-binding domains, in the proline-rich region and carboxy-terminal region of Tau. A total of more than 20 protein kinases can phosphorylate Tau proteins. This includes four groups of protein kinases: (1) the proline-directed protein kinases (PDPK), which phosphorylate Tau on Serines or Threonines that are followed by a Proline residue. This group includes cyclin-dependant kinase 2 and 5 (42–44), MAPK (45) and several SAPKs (46, 47). (2) The non-PDPK group includes Tau-tubulin kinases 1 and 2 (48–50), Casein kinases 1, 2 and 1δ (38, 51), DYRK1A (52–55), the phosphorylase kinase (56), Rho kinase (57), PKA, PKB/AKT, PKC, PKN (for review see (40)). (3) The third group includes protein kinases that phosphorylate Tau on Serine or Threonine residues followed or not by a Proline. Glycogen protein kinases (GSK3α and GSK3β) belong to this group and have a recognition motif (SXXXS or SXXXD/E) (58). This group also includes mitogen and stress-activated protein kinase MSK1, which belongs to the AGC kinases group. AGC kinases preferentially phosphorylate serine and threonine residues that lie in RXRXXS/T motifs (59). The S6 kinases p70 and p90 (RSK1 and RSK2) also phosphorylate Tau as well as serum- and glucocorticoid-induced kinase-1 (SGK1) (59). (4) The fourth group corresponds to tyrosine protein kinases such as Src kinases, C-abl and c-met (60) (see Diane Hanger Web site http://cnr.iop.kcl.ac.uk/hangerlab/tautable).
4 Tau Pathology
4.1 Tau in Neurofibrillary Degeneration
Neurofibrillary degeneration, as first described by Aloïs Alzheimer, consists of the intraneuronal accumulation of proteinaceous fibrils forming flame-shaped neurons. Fibrils were later demonstrated to correspond to paired helical filaments (PHF) indicating the well-structured organization of the molecular constituent (61). This material is insoluble and this biochemical property was used to isolate the material and to generate antibodies. The major antigen of PHF was shown to correspond to Tau protein, namely PHF-Tau (for review see (62)). Similarly, antibodies raised against isolated PHF-stained Tau proteins (63). Proteomic isolation and characterization of the most aggregated components of PHF showed that the core of PHF was mostly composed of the microtubule-binding motif lacking the exon 10 encoding sequences (64–66).
The most striking difference between postmortem Tau and PHF-Tau proteins is the molecular weight, as Tau protein-derived from postmortem control individual are resolved as six main bands (45–67 kDa), whereas PHF-Tau comprise four main bands between 60 and 74 kDa. Two-dimensional gel electrophoresis followed by Western-blotting is a useful method to demonstrate that PHF-Tau is more acidic than normal Tau (67, 68). Increased Tau acidity is due to hyper- and abnormal phosphorylation in degenerating neurons. A number of studies, using phosphatase, antibodies against PHF-Tau – thereafter showed to be phospho-dependant Tau antibodies – and mass-spectrometry analyses, together demonstrated that PHF-Tau is hyperphosphorylated on “native” phospho-sites and abnormally phosphorylated on pathologically related sites. The former observation was demonstrated using phospho-dependant antibodies and two-dimensional gel electrophoresis on human brain tissue obtained from tumor resections and, therefore, without postmortem/surgery delay (69, 70). Abnormal phospho-sites were demonstrated using antibodies that recognize PHF-Tau and not normal or “native” Tau, such as AT100 or AP422 that recognize the phosphorylated Threonines 212–214 and Serine 217, and the Serine 422, respectively. Nonetheless, to generate the pathological phospho-sites at AT100 epitope a sequential phosphorylation by two kinases (71) is necessary suggesting that several kinases may be deregulated in AD.
In AD disease, Tau pathology is found in brain areas affected with aging and several additional brains areas are also affected. However, the Tau pathology is following cortico-cortical connections with a stereotyped, sequential and hierarchical pathway affecting large pyramidal neurons. Braak and Braak proposed a six-stage spatiotemporal progression of Tau pathology in AD (72). A similar scheme of spatiotemporal spreading into ten stages is also described according to the brain regions sequentially affected: transentorhinal cortex (S1), entorhinal (S2), hippocampus (S3), anterior temporal cortex (S4), inferior temporal cortex (S5), mid-temporal cortex (S6), polymodal association areas (prefrontal, parietal inferior, temporal superior) (S7), unimodal areas (S8), primary motor (S9a) or sensory (S9b, S9c) areas and all neocortical areas (S10). Up to stage 6, the disease could be asymptomatic. In all the cases of our study, stage 7 individuals with two polymodal association areas affected by Tau pathology were cognitively impaired.
Yet, the cortical factors that trigger the cortical-spreading of Tau pathology in AD remain ill-defined (73–75). However, recent data suggest that neurofibrillary degeneration cortical spreading could follow a transmissible prion-like process. In fact, purified aggregates of PHF-Tau were purified from transgenic mice. Intracranial injection of this preparation was done in a mouse model, which overexpresses human Tau protein but does not display Tau pathology. Following the injection, development of a Tau pathology was observed. This pathology progressed from the injection site to neighboring brain structures (76).
5 Inherited Frontotemporal Dementia Linked to Chromosome 17 (FTDP) and Tau Gene Mutations
Historically, frontotemporal dementia (FTD) was often classified as a form of Pick’s disease, even when Pick cells or Pick bodies were not found (77). However, this denomination may involve different subgroups of pathologies, and the Lund and Manchester groups published in 1994 a consensus on Clinical and Neuropathological Criteria for Frontotemporal Dementia (1994). This publication clarified the position of Pick’s disease within FTD, and several of the reported cases of familial Pick’s disease were probably cases of familial FTD. Indeed it is difficult to ascertain families that have the classic pathological features of Pick’s disease from the literature (78), because they often have unusual clinical features.
In 1994, Wilhelmsen and colleagues described an autosomal dominantly inherited disease related to familial FTD, characterized by adult-onset behavioural disturbances, frontal lobe dementia, parkinsonism, and amyotrophy (79). They demonstrated a genetic linkage between this pathology, designated disinhibition–dementia–parkinsonism–amyotrophy complex (DDPAC) and chromosome 17q21–22 (79). Since then, several families sharing strong clinical and pathological features and for which there is a linkage with chromosome 17q22–22 have been described (80–83). They have been included in a group of pathologies referred to as frontotemporal dementia with parkinsonism linked to chromosome 17 (FTDP-17) (84).
Although clinical heterogeneity exists between and within the families with FTDP-17, usual symptoms include behavioural changes, loss of frontal executive functions, language deficit, and hyperorality. Parkinsonism and amyotrophy are described in some families, but are not consistent features. Neuropathologically, brains of FTD patients exhibit an atrophy of frontal and temporal lobes, severe neuronal cell loss, gray and white matter gliosis, and a superficial laminar spongiosis. One of the main characteristics is the filamentous pathology affecting the neuronal cells, or both neuronal and glial cells in some cases. The absence of amyloid aggregates is usually established (84, 85).
FTDP-17 has been related to mutations on the Tau gene (86–89). Tau mutations always segregate with the pathology and are not found in the control subjects, suggesting their pathogenic role. A total of 44 mutations are indexed. Thirty one missense mutations in coding regions, three silent mutations L284L, N296N and S305S, two single amino acid deletions ΔK280 and ΔN296 and nine intronic mutations in the splicing region following exon 10 at position +3, +11, +12, +13, +14, +16, +19, +29 and +33 have been reported (for review see http://www.molgen.ua.ac.be/FTDmutations). More recently, a Tau gene deletion encompassing exons 6 to 9 has been described in a French family and associated with the development of FTD (90).
Depending on their functional effects, Tau protein mutations may be divided into three groups: mutations affecting the alternative splicing of exon 10, and leading to changes in the proportion of 4R- and 3R-Tau isoforms, and mutations modifying Tau interactions with microtubules and those facilitating aggregation. Mutations of the Tau gene and their involvement in FTDP-17 emphasize the fact that abnormal Tau proteins may play a central role in the etiopathogenesis of neurodegenerative disorders, without any implication of the amyloid cascade. The functional effects of the mutations suggest that a reduced ability of Tau to interact with microtubules may be upstream of hyperphosphorylation and aggregation. These mutations may also lead to an increase in free cytoplasmic Tau (especially the 4R-Tau isoforms), and therefore facilitating their aggregation into filaments (91). Finally, some mutations may have a direct effect on Tau fibrillogenesis (92).
6 Tau Transgenic Mice
6.1 Tau-Knockout Mice
Tau expression was suppressed in different mouse models by MAPT deletion or invalidation (93–95). All of them appear physically normal and are able to reproduce. They do not display any change in central or peripheral nervous systems (94, 95). In one model, slight changes were seen in axonal diameter (93) and motor and cognitive deficits were also reported (96). Moreover, delay in axonal maturation was also reported in primary neuronal cell cultures from Tau-deficient mice (94). Tau deficiency is likely to be compensated by other microtubule-associated proteins such as MAP1A.
6.2 Wild-Type Tau Transgenic Mice
When 3R Tau isoforms are overexpressed in transgenic mice, hyperphosphorylated Tau mainly accumulates in spinal cord neurons. Main pathological observations were axonal degeneration, diminished number of microtubules, and reduced axonal transport (97, 98). Similar data were observed in transgenic mice expressing the longest human brain Tau isoform under the control of the human Thy-1 promoter. Hyperphosphorylated human Tau protein was present in nerve cell bodies, axons and dendrites (99–101).
Interestingly, transgenic mice bearing the human MAPT mini-gene were also developed (102). This model overexpresses human Tau proteins two- to threefold higher than murine Tau, but it does not exhibit any Tau pathology. However, surprisingly, when this model was crossbred with the murine MAPT gene deletion background model (95), the offspring displayed Tau pathology in a time-dependent manner, suggesting that murine Tau proteins may act as inhibitors of Tau aggregation (103). Independently, crossbreeding between other Tau-deficient mice (94) and transgenic mice with MAPT mini-gene was also used for understanding Tau splicing in early developmental stages (from 3R to 4R isoforms) (104). There is no data yet available on aged animals in these strains.
6.3 Mutated Tau Transgenic Mice
With the discovery of FTDP-17 mutations on MAPT, numerous transgenic models using these mutations were developed. They all allow for the development of Tau pathology characterized by Tau aggregation and neurofibrillary degeneration. Tau transgene is under various promoters (2′,3′-cyclic nucleotide 3′-phosphodiesterase, CaMKII, PDGF, Prion or Thy1.2) under an inducible system or not.
They display various phenotypes, with the most prominent one being motor deficits. In Table 1, we summarize the general phenotype of these models. In the next section, we describe four representative mutated Tau transgenic mice: rTg4510, TauRD/ΔK280, K3 and Thy-Tau22105–108, which allow for better understanding of Tau pathology.
Table 1
Main Tau transgenic mouse models
Strain name | Promoter | Mutation Tau construct | Phenotype | References |
|---|---|---|---|---|
P301S Tau | Thy1.2 | P301S 2-3-10+ | Neuron, motor | Allen et al., 2002 (116) |
]T-279 | MAPT | N279K 2+3+10+ | Neuron, glial, motor | Dawson et al., 2007 (117) |
]pR5 | Thy1.2 | P301L 2+3+10+ | Neuron, cogn | Götz et al., 2001 (118) |
](line 12) Pl Tg | 2′,3′-cyclic nucleotide 3′-phosphodiesterase | P301L 2+3-10+ | Glial, motor | Higuchi et al., 2005 (119) |
]K3 | Thy1.2 | K369I 2+3-10+ | Neuron, motor, park | Ittner et al., 2008 (107) |
]TauV337M | Thy1 | V337M 2+3+10- | Neuron, Cogn | Lambourne et al., 2007 (120) |
]Thy-Tau30 | Thy1.2 | G272V/P301S 2+3-10+ | Neur, motor | Leroy et al., 2007 (111) |
]JNPL3 | PrP | P301L 2-3-10+ | Neuron, Motor | Lewis et al., 2000 (121) |
]VLW | Thy1 | G272V P301L R406W | Neuron | Lim et al., 2001 (122) |
]TauRD/ΔK280 Proaggregation | CaMKII-driven rTA+tetOp (binary system) | ΔK280, 4R construct | Neuron, motor | Mocanu et al., 2008 (108) |
]TauRD/ΔK280/2P Antiaggregation | CaMKII-driven rTA+tetOp (binary system) | ΔK280/I277P/I308P, 4R construct | 4R construct expression | Mocanu et al., 2008 (108) |
]Tg-TauP301L | PrP | P301L 2+3+10+ | Neuron, glial, cogn | Murakami et al., 2006 (123) |
]rTg4510 | (inducible) CaMKII-driven rTA+tetOp (binary system) | P301L 2-3-10+ | Neuron, cogn, motor | Santacruz et al., 2005 (105) |
]Thy-Tau22 | Thy1.2 | G272V/P301S 2+3-10+ | Neuron, cogn | Schindowski et al., 2006 (106) |
]Tg214 | PDGF | V337M 2+3+10+ (myc/FlAG-tagged) | Neuron, cogn | Tanemura et al., 2001 (124) |
]R406W Tg | CaMKII | R406W 2+3+10+ (myc/FlAG-tagged) | Neuron, cogn | Tatebayashi et al., 2002 (125) |
In rTg4510 Tau transgenic mice, P301L mutant Tau protein is expressed in an inducible way. Following P301L Tau expression, these mice develop NFTs, neuron loss and behavioural impairment in a time-dependent manner (105). However, it is possible to switch off the expression of the transgene at a given time. Suppression of P301L Tau expression in rTg4510 Tau transgenic mice, which normally express the mutant protein at a high level, reverses behavioural impairments in these mice, although NFT formation continues. It suggested that NFT formation could be dissociated from neuronal dysfunction. In fact, soluble Tau rather than NFTs may be neurotoxic (105). Thus, NFT are unlikely to be the major toxic Tau species, at least in the early stages of Tauopathy. Two forms of Tau multimers (140 and 170 kDa), whose molecular weight suggests an oligomeric aggregate, were recently described in rTg4510 Tau transgenic mice. They accumulate early in the pathogenic cascade and their levels correlated consistently with memory loss at various ages in the rTg4510 mouse model (109). These Tau oligomers were also found in the brains of JNPL3 mice and patients presenting with FTDP-17 (109).
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


