Fig. 1
Flow graph of the described metagenomic-based approach to detect potential pathogens
3.1 Sample Preparation
1.
For each hive, collect approximately 50 adult worker bees in total, from both inside and outside the hive to ensure the presence of young and adult bees, in sterile containers. Keep frozen during transport at −18 °C or lower.
2.
Out of the collected material from a hive, manually homogenize 20 whole bees in a 30 ml glass homogenizer with 6 ml of 1× PBS. If possible, perform the homogenization on ice.
3.
Pour the homogenate into a 15 ml tube (or use several 2 ml tubes) and centrifuge at 1,000 × g for 10 min at 4 °C to pellet debris.
4.
Use a 5 ml syringe and a 0.45 μm PVDF syringe filter to remove any remaining debris. Draw a small amount of air (about 1 ml) into the syringe before filling with the supernatant. Attach the syringe filter and pass the supernatant through the filter into 1.5 ml tubes by applying a steady pressure. Use the air in the syringe to purge the filter. This will minimize fluid retention within the filter device and maximize sample recovery.
3.2 Enrichment by Nuclease Treatment and Nucleic Acid Extraction
1.
Aliquot 168 μl of the filtered homogenate into two 1.5 ml tubes and add 20 μl of 10× DNase buffer to each. Mix by vortexing and spin down briefly using a desktop mini centrifuge.
2.
To each tube, add 10 μl DNase I (10 U/μl) and 2 μl RNase (1 μg/μl). Mix by pipetting.
3.
Incubate at 37 °C for 2 h using a heat block or water bath.
4.
Dry the tubes, if necessary, and put them on ice. Store at −80 °C or proceed directly.
DNA Extraction
1.
Use the QIAamp DNA Mini Kit according to the manufacturer’s spin protocol for blood and body fluid DNA extraction (see Note 2 ).
2.
Measure DNA concentration by using a Qubit 2.0 Fluorometer with the dsDNA HS (High Sensitivity) Assay Kit, or use a NanoDrop ND-1000 to make an estimate, according to the manufacturers’ instructions. Concentrations are expected to range between 2 and 25 ng/μl.
3.
Aliquot the DNA and mark the tubes with the sample ID and date of extraction. Store at −80 °C.
RNA Extraction
The following steps should be carried out under a fume hood due to phenol component.
1.
Add 600 μl of TRIzol LS to each nuclease-treated homogenate and pipette the homogenate up and down a couple of times.
2.
Incubate the tubes at room temperature for 5 min.
3.
Add 160 μl of chloroform.
4.
Cap the tube securely and shake and/or vortex it vigorously for 15 s, so the contents of the tube get whitish.
5.
Incubate the tube for 2–3 min in room temperature.
6.
Centrifuge the tubes at 12,000 × g for 15 min at 4 °C. The rest of the centrifugation steps should be carried out at room temperature.
7.
Carefully take the tubes to the fume hood. There will be three phases after the centrifugation: an upper, colorless, aqueous phase containing RNA, a white interphase containing DNA, and a lower, red, organic phase.
8.
Carefully transfer the aqueous phase to a fresh 1.5 ml tube. Transfer it in small volumes without getting any of the other two phases into the pipette tip.
9.
Estimate the volume of the aqueous phase. Add the same volume of 70 % ethanol. Mix thoroughly by vortexing. Do not centrifuge.
10.
Pipet up to 700 μl of the sample into an RNeasy Mini Spin Column, or similar, in a 2 ml collection tube. Mark the lid of the column with the sample ID.
11.
Centrifuge at ≥8,000 × g for 15 s at 15–25 °C. Discard the flow-through. If there is still some of the sample left, transfer it to the Mini Column and centrifuge again.
12.
Proceed according to the manufacturer’s instructions.
13.
Measure the concentration of the extracted RNA by using a Qubit 2.0 Fluorometer with the RNA Assay Kit or control the RNA quality by using a 2100 Bioanalyzer with a RNA Pico Chip (Agilent Technologies) according to the instructions of the manufacturers. Concentrations are expected to range between 2 and 25 ng/μl.
14.
Aliquot the RNA and mark the tubes with the sample ID and date of extraction. Store at –80 °C.
3.3 Sequence-Independent, Single-Primer Amplification (SISPA)
Tag Labeling DNA
1.
Prepare the reagent mixture described in Table 1 in a 0.2 or 0.5 ml tube.
Table 1
Reagent mixture for tag labeling DNA
Component | Volume (μl) |
|---|---|
FR26RV-N (10 μM) | 2 |
DNA template | 10 |
dNTP mix (10 mM each) | 1.5 |
Exo(−) Klenow Buffer (10×) | 1.5 |
Total volume | 15 |
2.
Denature at 94 °C for 2 min in a thermal cycler and chill on ice for 2 min.
3.
Add 0.5 μl (2.5 U) of exo(−) Klenow DNA polymerase.
4.
Complete a first round of extension by incubating at 37 °C for 1 h. Proceed directly with denaturation at 94 °C for 2 min and chill on ice for 2 min.
5.
Add 0.5 μl (2.5 U) of exo(−) Klenow DNA polymerase for a second round of extension.
6.
Incubate at 37 °C for 1 h and inactivate the enzyme by incubating at 75 °C for 10 min.
7.
Tagged DNA fragments can be stored at −20 °C for several weeks.
Tag Labeling RNA
1.
Prepare the reaction mix 1 (Table 2) and 2 (Table 3) in separate 0.2 or 0.5 ml tubes.
Table 2
Reaction mix 1 for tag labeling RNA
Component | Volume (μl) |
|---|---|
FR26RV-N (10 mM) | 2 |
RNA template | 10 |
dNTP mix (10 mM each) | 1.5 |
Total volume | 13.5 |
Table 3
Reaction mix 2 for tag labeling RNA
Component | Volume (μl) |
|---|---|
Reaction mix 1 (see Table 2) | 13.5 |
RT buffer (5×) | 4 |
DTT (0,1 M) | 1 |
RNase inhibitor (40 U/μl) | 1 |
Reverse transcriptase (200 U/μl) | 1 |
Total volume | 20.5 |
2.
Keep reaction mix 2 on ice until step 4.
3.
Incubate reaction mix 1 at 65 °C for 5 min in a thermal cycler to denature the RNA.
4.
Chill on ice for 1 min and add reaction mix 2.
5.
Perform the reverse transcription by incubating at 25 °C for 5 min, 50 °C for 1 h, and 85 °C for 5 min. Place on ice.
6.
Add 0.5 μl (2.5 U) of exo(−) Klenow DNA polymerase for second-strand synthesis and incubate at 37 °C for 1 h. A final incubation at 75 °C for 10 min inactivates the enzyme.
7.
Tagged cDNA fragments can be stored at −20 °C for several weeks.
Amplification
1.
Perform the PCR amplification by using the complementary primer FR20RV. For each reaction, prepare the reaction mix described in Table 4 in a 0.2 or 0.5 ml tube.
Table 4
Reaction mix for PCR amplification
Component | Volume (μl) |
|---|---|
PCR buffer (10×) | 5 |
dNTP Mix (2.5 mM each) | 4 |
MgCl2 (25 mM) | 5 |
FR20RV (10 mM) | 4 |
DNA polymerase (5 U/μl) | 0.5 |
Nucleic acid template | 2.5 |
Nuclease-free H2O | 29 |
Total volume | 50 |
2.
Mix gently and, if necessary, spin down briefly using a desktop mini centrifuge.
3.
Perform the PCR in a thermal cycler with the conditions described in Table 5.
Table 5
Thermal cycling conditions
Cycle step | Temperature (°C) | Duration | Number of cycles |
|---|---|---|---|
Initial denaturation | 95 | 10 min | 1 |
Denaturation | 95 | 60 s | |
Annealing | 58 | 60 s | 40 |
Extension | 72 | 60 s | |
Final extension | 72 | 10 min | 1 |
4.
PCR products can be stored at −20 °C for several weeks.
Exonuclease Removal of Amplification Tag Sequences
1.
Purify the PCR products with the QIAquick PCR Purification Kit, according to the manufacturer’s instructions, or use a similar column-based purification system. Elute in 50 μl of EB buffer or water (pH 7.0–8.5).
2.
Use the restriction enzyme EcoRV to remove the amplification tag. Add purified PCR product, buffer, and restriction enzyme to a 1.5 ml microfuge tube as described in Table 6.
Table 6
Restriction reaction mixture
Component | Volume (μl) |
|---|---|
Purified PCR product | 26 |
Restriction enzyme buffer (10×) | 3 |
EcoRV (20 U/μl) | 0.5 |
Total volume | 29.5 |
3.
Incubate at 37 °C for 1 h using a heat block or water bath.
4.
Purify again using the QIAquick PCR Purification Kit or a similar column-based purification system. Elute in 50 μl of EB buffer or water (pH 7.0–8.5).
5.
To assess the compositions and concentrations of the final samples, use either a 2100 Bioanalyzer or run the samples on an agarose gel (1 % agarose in 0.5× TBE buffer) and measure the concentration using a Qubit 2.0 Fluorometer. Figure 2a, b illustrates successfully amplified random products when analyzed with the two abovementioned methods, respectively.
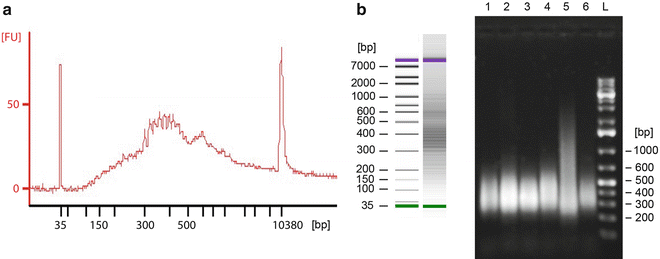
Fig. 2
Successfully amplified random products analyzed using (a) the 2100 Bioanalyzer DNA 1000 Assay and (b) separation on a 0.8 % agarose gel. The Bioanalyzer produces a graph where the y-axis represents fluorescence units [FU] and the x-axis is time, which translates to fragment size in base pair (bp). This information is also used to create a gel-like densitometry plot. On the agarose gel, sizes were determined using a 1 kb Plus DNA Ladder. A smear is expected due to the randomness of the amplification procedure and the DNA size typical range from 200 to 1,000 bp, with the mean size ~375 bp
3.4 Library Preparation and Sequencing
The generation of sequencing libraries as well as the actual sequencing is commonly performed by engaging a commercial provider or an academic core facility. This paragraph will therefore be kept brief and not outline a detailed protocol.
1.
Get Clinical Tree app for offline access

Since the preparation of sequencing libraries are platform specific, it is first necessary to select a sequencing platform. It has been demonstrated that HTS data in the range of 10–50 Mbp is enough to identify viruses with the described approach [24]. Most current platforms have a capacity in this range or higher. There are therefore other factors that might influence the choice, such as read length, cost (both per run and per Mb of DNA sequence), and ease of availability. For comparison, major commercial HTS platforms suitable for metagenomic-based detection and their characteristics are summarized in Table 7. A comprehensive review about HTS platforms has also been made by Glenn et al. [25].
Table 7




Characteristics and utility grades of HTS platforms suitable for metagenomic-based detection of potential pathogens
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree