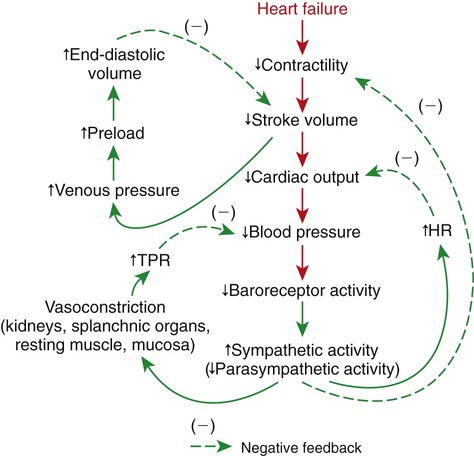
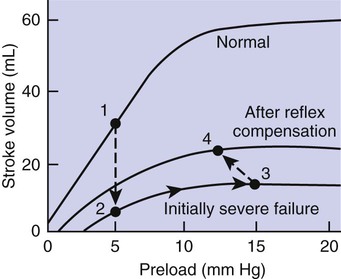
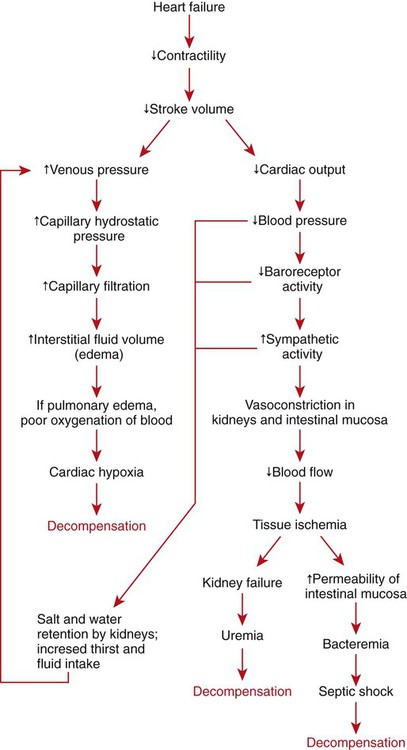
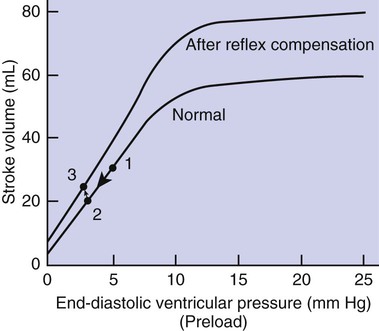
1. Both Starling’s mechanism and the arterial baroreflex help compensate for heart failure. 2. Serious complications secondary to heart failure include exercise intolerance, edema, salt and water retention, kidney failure, uremia, septic shock, and decompensation. 3. The immediate cardiovascular effects of hemorrhage are minimized by compensations initiated by the atrial volume receptor reflex and the arterial baroreceptor reflex. 4. The blood volume lost in hemorrhage is restored through a combination of capillary fluid shifts and hormonal and behavioral responses. 5. In large animals, the transition from a recumbent to a standing posture elicits the same cardiovascular responses as hemorrhage. 6. The initiation of exercise involves an interplay of local and neural changes that increases cardiac output and delivers increased flow to exercising muscle. Chapters 18 to 25 describe the various elements of cardiovascular function and control. An understanding of these individual elements is not sufficient, however, to provide a basis for the diagnosis and treatment of cardiovascular dysfunction. The veterinary clinician must understand the interaction of these elements in both normal and abnormal situations. Therefore this chapter discusses three fundamentally important, integrated cardiovascular responses: (1) the response to heart failure, (2) the response to hemorrhage, and (3) the response to exercise. In addition to elucidating important, integrated responses, this discussion provides a review and summary of key concepts of cardiovascular physiology. Ventricular function curves provide a helpful way to envision the consequences of heart failure and the compensations for heart failure. In Figure 26-1 the curve labeled Normal indicates the relationship between stroke volume and preload for a normal ventricle (for a review, see Figure 21-3, C). The curve labeled Initially severe failure shows that a ventricle in failure has a depressed contractility (i.e., a smaller stroke volume for any given preload). If a normal heart suddenly goes into severe failure, stroke volume changes from its normal value (shown by point 1) to the low value (shown by point 2). For purposes of illustration, imagine that these curves define the function of the left ventricle and that the left ventricle is the one that fails. A decrease in left ventricular stroke volume causes a decrease in left ventricular output, which results in a decrease in mean arterial blood pressure. If there is inadequate compensation for this fall in blood pressure, severe exercise intolerance is certain, inadequate perfusion of the critical organs is likely, and death is a strong possibility. However, several mechanisms react rapidly, within seconds to minutes, to compensate for heart failure and to minimize its adverse effects. One compensation for heart failure is Starling’s mechanism. If the left ventricle suddenly decreases its stroke volume, the right ventricle (at least for a few heartbeats) maintains a higher stroke volume than the failing left ventricle. The excess blood pumped by the right ventricle has to “go somewhere,” and most of the excess accumulates in the pulmonary veins and left atrium. In effect, blood backs up or dams up behind (i.e., upstream of) the left ventricle. The resulting increase in left atrial pressure creates an increase in left ventricular preload, which leads to an increase in left ventricular end-diastolic volume and, by Starling’s mechanism, an increase in stroke volume. This improvement in stroke volume is depicted in Figure 26-1 as a transition from point 2 to point 3. The sequence of events, whereby an increase in preload helps offset the initial fall in stroke volume, is also diagrammed in Figure 26-2 (top left loop). Note that the compensation by Starling’s mechanism does not return stroke volume to its normal value because contractility remains severely depressed; however, without this compensation severe heart failure would be fatal. The sympathetic effect on the heart increases ventricular contractility. Contractility is not restored to normal but is brought to a higher level than existed in the absence of reflex compensation. Graphically, the effect of the baroreflex is to move the failing ventricle to a function curve that is intermediate between the Normal curve and the curve of Initially severe failure (see point 4 in Figure 26-1). Note that the increase in contractility also brings stroke volume back toward (but not reaching) its normal level. The net effect of the compensations by way of Starling’s mechanism and the baroreflex is that arterial blood pressure can be maintained near its normal level, at least when the animal is at rest, despite a severe ventricular failure. Figure 26-2 summarizes these reflex effects. Note that after compensation by Starling’s mechanism and the baroreflex, contractility, stroke volume, cardiac output, and blood pressure remain at least somewhat below normal. By contrast, preload, sympathetic activity, heart rate, and TPR are above normal. Edema is another serious complication secondary to heart failure. As noted, blood dams up in the atrium and veins behind a failing ventricle. In the case of left ventricular failure, left atrial pressure increases, as does pressure in the pulmonary veins and pulmonary capillaries. The increase in pulmonary capillary hydrostatic pressure leads to an increase in the filtration of capillary fluid into the interstitial spaces of the lungs. Pulmonary edema develops. The excess of interstitial fluid slows the transfer of oxygen from the lung alveoli into the lung capillaries and can result in inadequate oxygenation of the blood (hypoxemia). In extreme cases, interstitial fluid leaks into the intrapleural space (pleural effusion) or into the alveolar air spaces, which causes a further reduction in lung function. The resulting hypoxia in critical organs can be fatal. In a patient with right ventricular failure the increase in venous pressure occurs in the systemic circulation. Therefore the resulting edema occurs in the systemic organs, particularly in dependent extremities and in the abdomen. The cause-and-effect sequence by which heart failure leads to edema is summarized in Figure 26-3 (top left). Whether the edema is in the lungs or in the systemic circulation, its degree is limited by the three safety factors previously discussed (see Figure 23-5). These safety factors would probably keep the edema of heart failure well controlled were it not for an additional factor that exaggerates the elevation of venous pressure in heart failure. As long as arterial pressure remains subnormal in a patient with heart failure, the baroreceptor reflex and some mechanisms involving the kidneys work to raise blood volume above normal. These volume-increasing mechanisms include increased thirst (which raises fluid intake), increased release of antidiuretic hormone (ADH) from the pituitary (which decreases the amount of fluid lost in the urine), and activation of the renin-angiotensin-aldosterone system (which decreases sodium loss in the urine). These effects of the baroreflex were mentioned briefly in Chapter 25; the mechanisms involving the kidneys are described in more detail in Chapters 41 and 43. The point for now is that the patient with severe heart failure experiences a substantial and persistent increase in blood volume. The excess blood accumulates particularly in the veins upstream from the failing ventricle, which exaggerates the increases in venous pressure and capillary filtration. The normal safety factors against may be overwhelmed. This is why one of the main goals in the clinical treatment of heart failure is to counteract the buildup of excessive blood volume and interstitial fluid volume. Diuretic drugs are the main therapies used for this purpose (see Chapter 43). Intense and prolonged vasoconstriction in the splanchnic circulation can also have lethal consequences. The mucosa of the gastrointestinal tract is particularly susceptible to ischemic damage. Normally, the intestinal mucosa creates a barrier between the contents of the intestinal lumen and the bloodstream. Ischemic damage to the intestinal mucosa allows bacteria and bacterial toxins to pass into the bloodstream or the peritoneum. The resulting bacteremia or peritonitis can cause septic shock and death. The causes and consequences of renal and splanchnic ischemia are summarized in Figure 26-3 (bottom right). Other decompensatory cycles develop in cases of severe, prolonged heart failure, but these three examples (which are illustrated in Figure 26-3) show why decompensation is such a serious development. Figures 26-4 and 26-5 summarize the cardiovascular responses to hemorrhage. The curve labeled Normal in Figure 26-4 shows that the maintenance of a normal stroke volume depends on the maintenance of a normal level of ventricular preload. When hemorrhage occurs, blood is lost from the whole cardiovascular system, but particularly from the veins, which are the blood reservoirs of the body. Hemorrhage therefore decreases venous volume, venous pressure, atrial pressure, ventricular preload, and ventricular end-diastolic ventricular volume. In the absence of any compensation, ventricular stroke volume decreases from point 1 in Figure 26-4 to point 2.
Integrated Cardiovascular Responses
Both Starling’s Mechanism and the Arterial Baroreflex Help Compensate for Heart Failure
Serious Complications Secondary to Heart Failure Include Exercise Intolerance, Edema, Salt and Water Retention, Kidney Failure, Uremia, Septic Shock, and Decompensation
The Immediate Cardiovascular Effects of Hemorrhage Are Minimized by Compensations Initiated by the Atrial Volume Receptor Reflex and the Arterial Baroreceptor Reflex
< div class='tao-gold-member'>
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Integrated Cardiovascular Responses
Only gold members can continue reading. Log In or Register to continue
