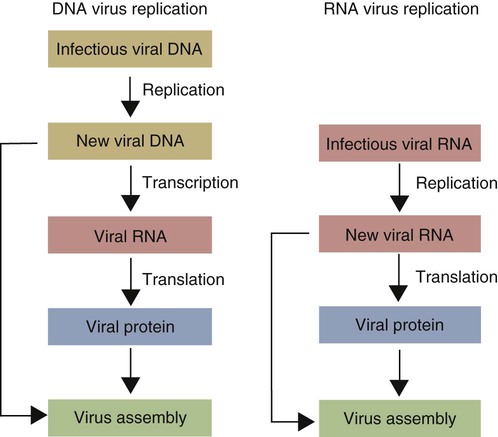
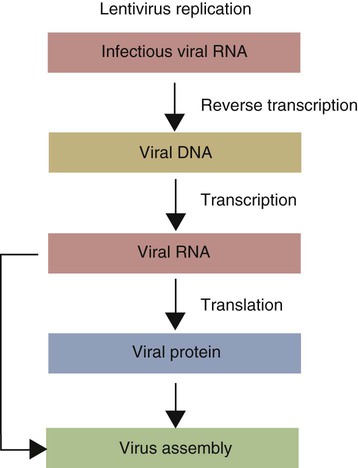
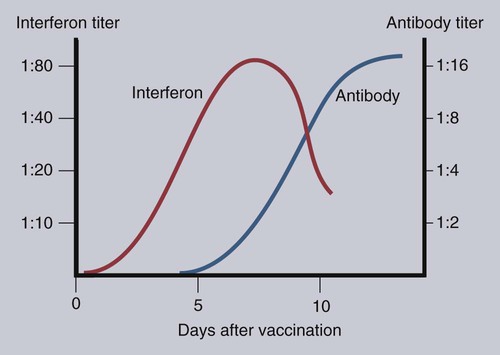
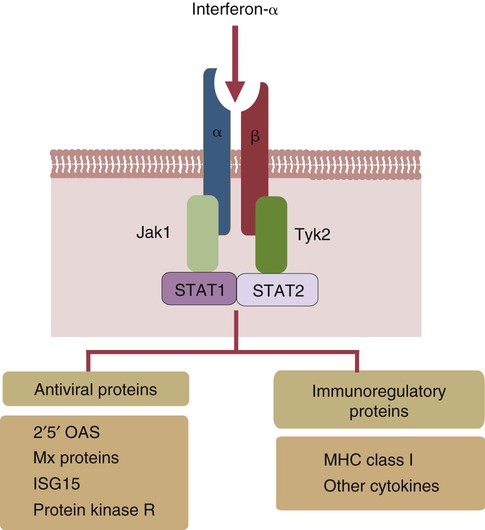
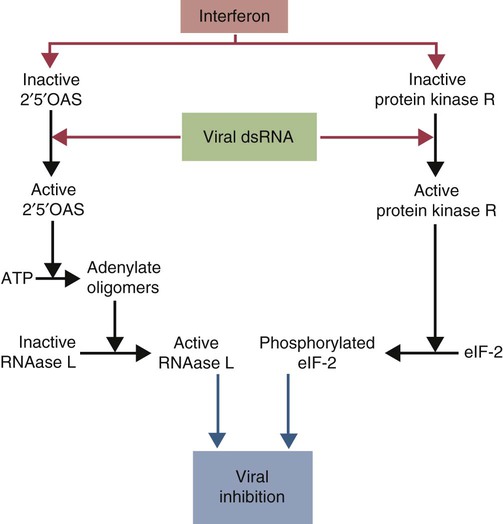
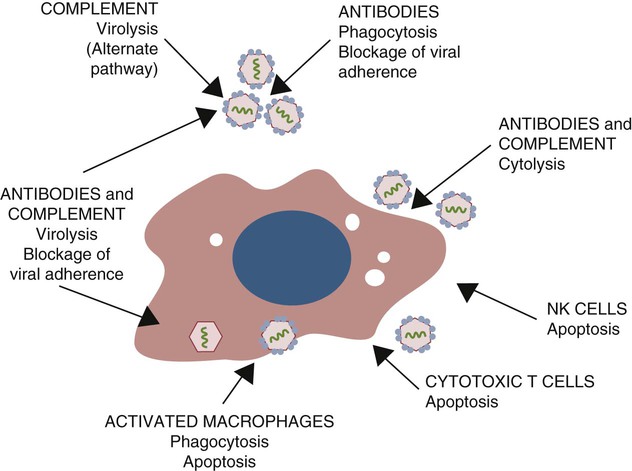
• When sentinel cells detect foreign nucleic acids through their TLRs and other receptors, they are triggered to secrete the antiviral type I interferons. • Antibodies are effective against extracellular viruses and can prevent them binding and infecting cells and thus neutralizing them. • Cell-mediated responses are primarily responsible for antiviral immunity. The major mechanism involved is the killing of virus-infected cells by cytotoxic T cells. • Because viruses are obligate intracellular parasites, they employ a wide variety of methods of evading the immune response. • Some viruses may cause minimal disease themselves, but significant damage and disease may be caused by the immune response to these viruses. In studying the nature of the host responses to viruses, it is well to recognize that this continuing selective pressure on both host and virus exists and profoundly influences the outcome of all viral infections (Box 26-1). Virus particles, called virions, consist of a nucleic acid core surrounded by a layer of proteins (see Figures 9-2 and 38-2). This protein layer, called the capsid, is made up of subcomponents called capsomeres. An envelope containing lipoprotein may also surround virions. The complexity of viruses varies. Some, such as poxviruses, are complex, whereas others, such as foot-and-mouth disease virus, are relatively simple. Antibodies can be produced against epitopes on all the proteins situated inside and on the surface of the virion. Antibodies against the nucleoprotein components are not usually significant from a protective point of view, but they may be useful for serologic diagnosis. Adsorption, the first step in the invasion of a cell by a virus, occurs when a virus binds to receptors on the cell surface. These receptors have not evolved for the convenience of viruses but have some other physiological function. The rabies virus binds to the receptor for acetylcholine, a neurotransmitter. The Epstein-Barr virus (the cause of infectious mononucleosis) binds to a receptor for C3. Rhinoviruses that cause the common cold bind to cell-surface integrins. The chemokine receptor CCR5 has been identified as the receptor used by West Nile virus. The nature, number, and distribution of host cell receptors determine the host range and tissue tropism of a virus. The bound virion is taken into the cell through endocytosis or by fusion with the plasma membrane. Once inside a cell, the capsid is dismantled so that its nucleic acid is released into the cell cytoplasm, a process called uncoating. Once the virus genome is uncoated, replication begins (Figure 26-1). The host cell DNA, RNA, and protein synthesis are usually inhibited so that only viral genetic information is processed. If the virus, for example, a herpesvirus, contains DNA, this viral DNA is replicated. The new viral DNA is then transcribed into viral messenger RNA (mRNA), and this RNA is translated into new capsid proteins. These new proteins are then assembled into virions. The host cell also replicates the viral nucleic acid so that large quantities of viral DNA are produced. The viral DNA is packaged inside the new capsids so that complete virions are formed. If the virus is unenveloped, the infected cells rupture, and the virions are released into the environment. If the virions are enveloped, they leave the cell by budding through the cell surface. The cell membrane that encloses them serves as the new envelope. The released virions may then spread to nearby cells and invade them in turn. A different replication mechanism is employed in the case of some RNA tumor viruses and immunodeficiency viruses (Figure 26-2). These are called retroviruses since their RNA is first reversely transcribed into DNA by an enzyme called a reverse transcriptase. The new viral DNA is then integrated into the host cell genome as a provirus. This proviral DNA can then be transcribed into RNA, as well as being able to copy itself. The proteins and RNA can then be packaged into a complete new virion. The interferons are cytokines that protect other cells against viral, bacterial, and protozoan invasion. They are all glycoproteins of 20 to 34 kDa. They are classified into three major types: I, II, and III. There are many type I interferons, each denoted by a greek letter. These include IFN-α, produced in large quantities by plasmacytoid dendritic cells and in much smaller amounts by lymphocytes, monocytes, and macrophages. Most mammals produce multiple isoforms of IFN-α. (There are 18 different isoforms in humans, 12 in pigs and cattle, 4 in horses, 2 in dogs). IFN-β is derived from virus-infected fibroblasts. (There are 5 isoforms in cattle and pigs and 1 in dogs and humans.) IFN-ω is produced by lymphocytes, monocytes, and human, horse, pig, rabbit, and dog trophoblast cells (6 to 7 in pigs, 5 in humans, 2 in horses, and none in dogs). A distinct form of type I interferon, IFN-τ, has been isolated from the ruminant trophoblast, and IFN-δ has been isolated from the pig trophoblast. IFN-δ is only distantly related to the other type I interferons. IFN-ε is a member of the type I family whose expression is limited to reproductive and brain tissues. It appears to play a role in mucosal and nervous system immunity. In most cases these molecules act on virus-infected cells to inhibit viral growth. The trophoblast interferons also regulate the maternal immune response to a fetus (see Figure 32-9). There is only one type II interferon, IFN-γ, a produced by antigen-stimulated T cells. It is also produced in pig trophoblast cells (Box 26-2). The two major type I interferons (IFN-α and IFN-β) are produced by virus-infected cells within a few hours after viral invasion, and high concentrations may be achieved in vivo within a few days, long before adaptive immunity develops. For example, in cattle infected intravenously with bovine herpesvirus-1 (BHV-1), peak interferon levels in serum are reached 2 days later and then decline, but they are still detectable by 7 days (Figure 26-3). In contrast, antibodies are not usually detectable in serum until 5 to 6 days after the onset of a virus infection. Type I IFN-α and IFN-β are secreted by virus-infected cells, and both bind to a heterodimeric receptor (IFNAR) on nearby cells activating a JAK/STAT signaling pathway (see Figure 8-8) (Figure 26-4). Receptor binding results in the development of an “antiviral state” within a few minutes that peaks 5 to 8 hours later. This virus resistance is mediated through four major pathways. • The 2′5′ A pathway: Type I interferons upregulate transcription of the genes coding for 2′5′-oligoadenylate synthetases (2′5′-OAS). Expressed OAS enzymes are then activated by exposure to dsRNA. The activated enzymes act on adenosine triphosphate (ATP) to form 2′5′ adenylate oligomers. These oligomers in turn activate a latent ribonuclease called RNAase L (Figure 26-5). RNAase L degrades viral RNA and inhibits viral growth. • The Mx guanosine triphosphatase (GTPase) pathway: Mx proteins are interferon-induced GTPases that accumulate as oligomers on intracellular membranes such as the smooth endoplasmic reticulum. Following viral infection, Mx monomers are released. These bind and trap viral nucleocapsids and other essential viral components and so block the assembly of new viruses. Mx proteins are expressed in many different cell types such as hepatocytes, endothelial cells, and immune cells. They inhibit a wide range of RNA viruses, including the influenza viruses. • The protein kinase R (PKR) pathway: PKR is induced by type I interferons. The inactive kinase accumulates in the cell nucleus and cytoplasm, where it is activated directly by viral RNAs. Activated PKR regulates several cell signaling pathways and phosphorylates an initiation factor called eIF2α, which then prevents translation initiation of viral mRNA. • The ISG15 pathway: One of the most prominent of the interferon-stimulated genes is ISG15. This codes for a 17-kDa, ubiquitin-like protein that binds to many different proteins and enhances their destruction. It is not known how this results in increased antiviral resistance and reduced viral replication. The ability of cells to produce interferons varies. Virus-infected leukocytes, especially plasmacytoid dendritic cells, produce large amounts of IFN-α; virus-infected fibroblasts produce IFN-β; and antigen-stimulated T cells are the major source of IFN-γ (Chapter 14). Kidney cells are poor interferon producers, and neutrophils produce no interferon. Lymphocytes from normal, unsensitized donors can kill virus-infected cells. This innate cytotoxicity is due to natural killer (NK) cells (Chapter 19). NK cell cytotoxicity is stimulated by type I interferons and, as a result, is important early in a virus infection. Indeed NK cells provide a first line of defense against many viruses. NK cells also produce IFN-γ, and this too has a direct antiviral effect. NK cells may therefore reduce the severity of viral infections long before the development of adaptive immunity and the appearance of specific cytotoxic T cells. Virus capsid and envelope proteins are antigenic, and it is against these that antiviral antibody responses are largely mounted (Figure 26-6). Antibodies can prevent cell invasion by blocking the adsorption of virions to target cells, by stimulating phagocytosis of viruses, by triggering complement-mediated virolysis, or by causing viral clumping and thus reducing the number of infectious units available for cell invasion. Binding of antibodies alone does not destroy viruses since the splitting of virus-antibody complexes may release infectious virions. Although antibodies and complement can neutralize free virions and destroy virus-infected cells, cell-mediated immune responses are much more important in controlling virus diseases. This is readily seen in immunodeficient humans (Chapter 37). Those who cannot mount an antibody-mediated response suffer from overwhelming bacterial infections but tend to recover from the common viral diseases. In contrast, humans with a T cell deficiency are commonly resistant to bacterial infection but highly susceptible to virus diseases. Macrophages develop antiviral activity following activation. Viruses are readily endocytosed by macrophages and are usually destroyed. If the viruses are noncytopathic but can, however, grow inside macrophages, a persistent infection may result. Under these circumstances, the macrophages must be activated to eliminate the virus. Thus immunity mediated by IFN-γ is a feature of some virus diseases (Chapter 18). For example, macrophages from birds immunized against fowlpox show an enhanced antiviral effect against Newcastle disease virus and will prevent the intracellular growth of Salmonella gallinarum, a feature that is not a property of normal macrophages.
Immunity to Viruses
Virus Structure and Antigens
Pathogenesis of Virus Infections
Innate Immunity
Interferons
Antiviral Activities
Adaptive Immunity
Antibody-Mediated Immunity
Cell-Mediated Immunity
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


