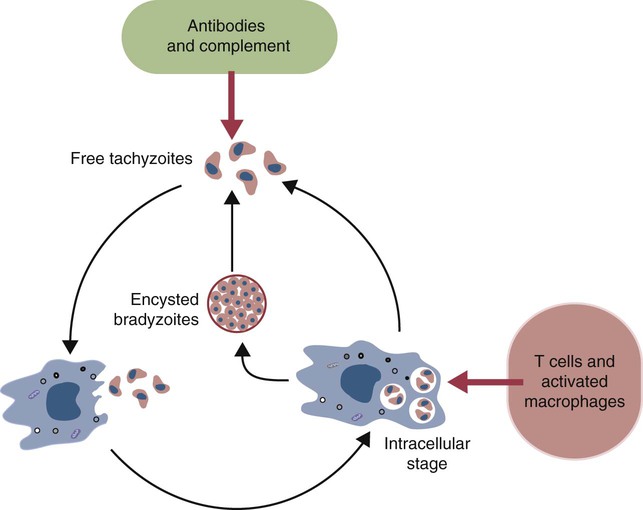
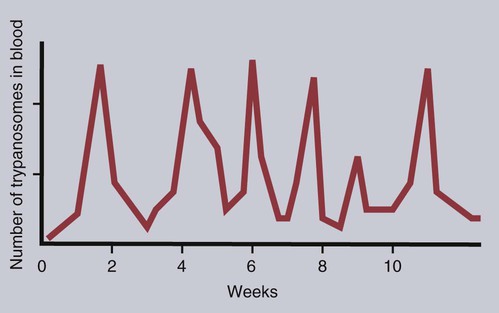
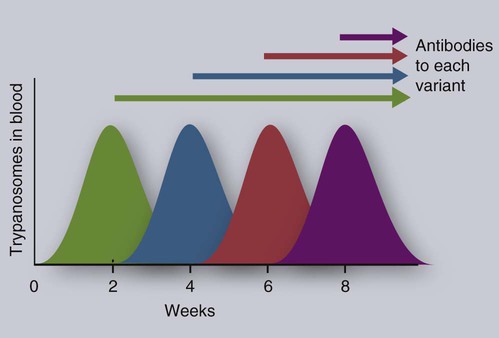
• Parasites, by definition, are able to evade their host’s immune response for at least sufficient time for the parasite to reproduce. • In general antibody-mediated immune responses protect against extracellular protozoa, whereas cell-mediated responses control intracellular protozoa. • Protozoan parasites employ some very sophisticated techniques to ensure their survival in the face of an animal’s immune response. • Helminth parasites have a unique ability to trigger Th2 responses and immunoglobulin E (IgE) production. IgE may have evolved as an antiparasite antibody. • Parasitic worms have a thick cuticle that protects them against damage caused by most protective cells. However, eosinophils appear to be uniquely able to damage and kill helminths. • Immunity to parasitic arthropods such as ticks and biting flies also appears to be a property of the Th2 responses. However, although immune responses can reduce arthropod feeding and reproduction, they rarely kill the arthropods. A consistent feature of all parasite infestations however, is that they block or significantly delay the innate and adaptive defenses of their host so that they may persist for sufficient time to reproduce. Some parasites may simply delay their destruction until they complete a single life cycle. Other well-adapted parasites may contrive to survive for the life of their host protected from immunological attack by sophisticated and specific evasive strategies (Figure 27-1). Intracellular parasites use many different and unique strategies to invade cells and inhibit intracellular killing. Most gain entry to a cell by employing host-mediated processes such as phagocytosis or induced uptake. Apicomplexans such as Toxoplasma and Cryptosporidium, however, actively penetrate cells using a system of adhesion-based motility called “gliding.” Once inside, these parasites reside in specially modified vacuoles. Protective immunity against apicomplexan protozoa, such as Cryptosporidium, Eimeria, Neospora, Plasmodia, and Toxoplasma species, is generally mediated by Th1 responses. For example, T. gondii is an obligate intracellular parasite whose tachyzoites grow within cells, especially macrophages (Figure 27-2). The parasites produce perforin-like molecules that permeabilize the cell membrane and permit the tachyzoites to escape and invade other cells. They penetrate these cells by “gliding” through a molecular junction in the cell membrane and do not trigger proper phagosome formation or maturation. Toxoplasma tachyzoites are therefore not destroyed since their “parasitophorous vacuoles” do not mature and fuse with lysosomes. Toxoplasma can grow inside cells in an environment free of antibodies, oxidants, or lysosomal enzymes. Toxoplasma-infected dendritic cells are attacked and killed by perforins from natural killer (NK) cells. However, the released parasites can then invade the NK cells! These infected NK cells, in contrast, are not effectively targeted by other NK cells. The production of IL-12 and IFN-γ is essential for the early control of Toxoplasma infection. The toxoplasma actin-binding protein profilin is the ligand for TLR11. TLR11 on dendritic cells signals through the MyD88 pathway stimulating IL-12 and IFN-γ production. T. gondii cyclophilin also stimulates IL-12 production from dendritic cells through CCR5. The IL-12 and IFN-γ in turn trigger a strong Th1 response. Some antibodies are produced that, together with complement, destroy extracellular organisms and prevent its spread between cells (Figure 27-3). This response, however, has little or no influence on the intracellular forms of the parasite. The intracellular organisms are only destroyed by the IL-12-dependent Th1 response. Activated Th1 cells secrete IFN-γ in response to Toxoplasma ribonucleoproteins. This IFN-γ activates macrophages, permitting lysosome-vacuole fusion and killing the intracellular organisms. In addition, cytotoxic T cells can destroy Toxoplasma tachyzoites and Toxoplasma-infected cells on contact. T. gondii tachyzoites, however, may convert into a cyst form containing bradyzoites. The cysts are weakly immunogenic and do not stimulate inflammation. It is possible that this cyst stage is not recognized as foreign. As a result, the cysts persist indefinitely within tissues. In Theileria parva infection (East Coast fever) of cattle, sporozoites can invade α/β and γ/δ T cells, as well as B cells. The parasite then activates NF-κB by continuously phosphorylating its inhibitor proteins Iκ-Bα and Iκ-Bβ (Chapter 8). The NF-κB thus persists, maintains the cell in an activated state, and prevents its apoptosis. The activated cells produce both IL-2 and IL-2R. As a result, a loop is established, by which infected cells secrete IL-2, which in turn stimulates their growth. As Theileria schizonts develop within lymphocytes, the infected cells enlarge and proliferate. Since the parasite divides synchronously with its host cell, there is a rapid increase in parasitized cells resulting in overwhelming infection and death. Some animals, however, may recover from infection and become solidly immune. In these animals, CD8+ T cells can kill infected lymphocytes by recognizing parasite antigens in association with MHC class I molecules. In susceptible animals, the parasites interfere with MHC class I expression. The clinical signs of leishmaniasis are directly linked to the immune response of the infected dog. In susceptible animals, the organisms may spread from the skin to the local lymph node, spleen, and bone marrow within a few hours. In resistant dogs, the parasites remain restricted to the skin and draining lymph node and either remain healthy or develop a mild, self-limited disease. These resistant dogs mount a weak antibody response but a strong and effective Th1 response. They may have low antibody titers, but they produce IFN-γ in response to parasite antigens, generate type I granulomas, mount strong delayed hypersensitivity responses, and eventually destroy the parasites. Resistance to Leishmania has a strong genetic component; for example, Ibizian hounds appear to be resistant to this parasite. There is also an association between resistance and certain MHC class II alleles as well as some Slc11a1 (Nramp) alleles in dogs (see Box 6-1). Although reduced antigenicity may be considered the ultimate evasive technique, many protozoa, especially the trypanosomes, successfully employ repeated antigenic variation. If cattle are infected with the pathogenic trypanosomes T. vivax, T. congolense, or T. brucei and their parasitemia measured at regular intervals, the numbers of circulating organisms are found to fluctuate greatly. Periods of high parasitemia alternate regularly with periods of low or undetectable parasitemia (Figure 27-4). Serum from infected animals contains antibodies against trypanosomes isolated before bleeding but not against those that develop subsequently. Each period of high parasitemia corresponds to the expansion of a population of trypanosomes with a new surface glycoprotein antigen. The elimination of this population by antibodies leads to a rapid fall in parasitemia. Among the survivors, however, some parasites express new surface glycoproteins and grow without hindrance. As a result, a fresh population arises to produce yet another period of high parasitemia (Figure 27-5). This cyclical fluctuation in parasite levels, with each peak reflecting the appearance of a new population with new surface glycoproteins, can continue for many months. The major surface antigens of these trypanosomes are known as variant surface glycoproteins (VSGs). These are the antigens targeted by host antibodies. The VSGs produced early in trypanosome infections tend to develop in a predictable sequence. However, as the infection progresses, the production of VSGs becomes more random. The VSGs form a thick coat on the surface of the trypanosome. When antigenic change occurs, the VSGs in the old coat are shed and replaced by an antigenically different VSG. Analysis indicates that these trypanosomes possess about 200 VSG genes, with an additional 1600 silent genes, of which two thirds are pseudogenes. Antigenic variation occurs as a result of repeated DNA breaking and repair, replacing an active VSG gene with one from the silent gene pool. Since only a small part of the tightly packed VSG is exposed to host antibodies, it is not even necessary for the complete molecule to change. Replacement of exposed epitopes by gene conversion is sufficient for effective variation (Chapter 17). Early in infections, complete VSG gene replacement occurs. Later on, partial replacement and point mutations can create new antigenic specificities. In some cases, the expressed VSG gene can be constructed as a mosaic from several archival pseudogenes. The potential for recombination-based variation is therefore absolutely enormous. The immune responses against protozoa may cause hypersensitivity reactions that contribute to disease. Type I hypersensitivity is a feature of trichomoniasis and results in local irritation and inflammation in the genital tract. Type II cytotoxic reactions are of significance in babesiosis and trypanosomiasis, in which they contribute to the anemia. In babesiosis, red cells express parasite antigens on their surfaces and are thus recognized as foreign and eliminated by hemolysis and phagocytosis. In trypanosomiasis, either fragments of disrupted organisms or possibly preformed immune complexes bind to red cells and provoke their elimination, causing anemia. Immune complex formation on circulating red cells is not the only problem of this type in trypanosomiasis. In some cases, excessive immune complex formation can lead to vasculitis and glomerulonephritis (type III hypersensitivity; see Chapter 30). Immune complex lesions are a marked feature of visceral leishmaniasis, as described previously. Trypanosome infections may trigger an enormous increase in IgM-secreting cells so that very high levels of IgM are found in the blood of infected animals. Some of these antibodies are directed against autoantigens. These include rheumatoid factor–like molecules, and antibodies against thymocytes, single-stranded DNA, red cells, and platelets. In T. congolense–infected cattle, these polyclonally stimulated B cells are BoCD5+. As pointed out earlier, CD5+ B1 cells are of a different lineage to conventional B2 cells (Chapter 15). The mechanism of this polyclonal B cell activation is unknown. It is probable that a type IV hypersensitivity reaction contributes to the inflammation that occurs when Toxoplasma cysts break down and release fresh tachyzoites. Extracts of T. gondii (toxoplasmin), if administered intradermally to infected animals, will cause a delayed hypersensitivity response (Figure 27-6).
Immunity to Parasites
Immunity to Protozoa
Innate Immunity
Adaptive Immunity
Leishmaniasis
Evasion of the Immune Response
Adverse Consequences
< div class='tao-gold-member'>
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Immunity to Parasites

Only gold members can continue reading. Log In or Register to continue
