and Damian C. Crowther1, 2
(1)
Department of Genetics, University of Cambridge, Cambridge, UK
(2)
Department of Medicine, University of Cambridge, Cambridge Institute for Medical Research, Cambridge, UK
Abstract
In the quest for understanding human neurodegenerative disorders, a variety of organisms have been used to create disease models. Because of its many advantages, Drosophila melanogaster is currently being used to model many human conditions including poly Q expansion disorders such as Huntington’s disease, Parkinson’s disease, Alzheimer’s disease (AD), and other dementias. AD is characterized by two pathologies; the first, extracellular amyloid β (Aβ) plaques, consist mainly of toxic Aβ42 peptide, and the second, intracellular neurofibrillary tangles, consists mainly of hyperphosphorylated tau protein. Drosophila AD models have focused either on replicating the amyloid precursor protein (APP) processing model (Aβ is a proteolytic product of APP) or on expression of simpler secreted Aβ peptides in the fly nervous system. These models replicate many of the features of human AD, including Aβ deposition, neuronal loss and neurodegeneration, and behavioral phenotypes such as impaired learning and memory. In recent years, following the characterization of these models, focus has shifted to utilizing the genetic power of Drosophila and screens are being conducted for identifying modifiers of AD pathology.
Key words
Alzheimer’s disease Drosophila melanogaster fruit flydementiasurvivallocomotorgenetic screensmodifiers1 Introduction
1.1 Why Use the Fruit Fly to Model Neurodegenerative Diseases?
The devastating symptoms of neurodegenerative diseases, such as movement disorders, ataxia, tremor, and cognitive and memory loss, are caused by neuronal dysfunction and loss. Many of these diseases are caused by aberrant processing and accumulation of proteins in the brain. To model these diseases in the fruit fly Drosophila melanogaster , we require some pre-existing knowledge about the pathology of the disease. In many cases, this understanding has come from studying families in which a normally sporadic disease is inherited as a dominant trait (1). The findings from studying such families have uncovered pathological events that are sufficient to cause the disease. In Alzheimer’s disease (AD) for instance, where only 10% of cases are familial, all the mutations uncovered (2) involve the proteolytic processing of the amyloid precursor protein (APP) and the generation of the amyloid-β peptide (Aβ). Since the symptoms of patients with familial and sporadic AD are very similar (3), it makes sense to use the insights from studying familial AD mutations in order to recapitulate AD in model organisms.
For an organism to be a faithful model of disease, it is necessary that the basic aspects of cell biology in this organism are conserved in higher-order metazoans (i.e. humans). Indeed, it has been shown that 75% of human disease-linked loci have homologues in Drosophila (4). It is also essential that the basic developmental programs are conserved between fruit flies and humans. In fact, many signaling pathways conserved among metazoans such as the wnt pathway were first identified and characterized in Drosophila (5), and subsequently shown to be crucial in humans as well (6). The Drosophila brain, despite being small, is amenable to study, as it comprises more than 300,000 neurons and is divided into compartments with specialized functions such as learning, memory, olfaction, and vision. In addition, fruit fly researchers have made available a plethora of genetic tools, including large collections of mutants (7) and transposon-based methods for generation of transgenics, which when coupled with a well-annotated genome and limited genetic redundancy allow for large genetic screens to identify genetic interactors (8). Finally, compared to mice, Drosophila are small, fecund, cheap-to-maintain organisms, with a short generation time, which allows us to quickly generate results that can then be exploited for experimental design in higher organisms.
1.2 Fly Models of Neurodegenerative Diseases
Drosophila can be used to model human diseases in many different ways. In some cases, researchers have investigated the Drosophila homologue of a human disease, in order to examine the normal function of the disease-linked protein. This approach has been used in examining, for instance, the role of the Drosophila homologues of the AD-associated genes, APP (APPL in the fly) (9,10) and presenilins 1 and 2 (PS1 and PS2, DPsn in the fly) (11), uncovering roles for APPL and DPsn in axonal transport and notch signaling, respectively. Alternatively, since most human neurodegenerative diseases are caused by autosomal dominant mutations or the abnormal accumulation of a protein, a model system may be generated by overexpressing the human disease gene (or a disease-causing mutant) or its Drosophila homologue. Such an approach has been used in modeling diseases caused by tau phosphorylation (12), α-synuclein (13), and various polyQ-containing proteins (14,15) that are involved in tauopathies, Parkinson’s disease (PD), and polyQ-diseases such as Huntington’s disease (HD), respectively. Neurodegenerative diseases can also be studied in fruit flies by unbiased, forward genetics approaches, where flies are mutagenized and then screened for certain phenotypes such as behavioral defects or decreased longevity. Such screens have identified interesting mutants such as bubblegum (16), eggroll, and spongecake (17), and have identified genes and processes potentially involved in human diseases with similar pathology (for review see (18)).
The first fly model of neurodegenerative disease was a model of spinocerebellar ataxia type 3 (SCA3, one of the coding and noncoding CAG expansion (polyQ) diseases) (14). Various faithful models for many polyQ diseases have since been generated by expression of human polyQ-containing proteins, with varying numbers of CAG repeats. These models for both ataxins (causing different SCAs) and for Huntingtin have shown progressive neurodegeneration and phenotype severity that correlates with the number of CAG repeats ((14,15), for review see (19,20)). The fly models for HD and the various ataxias have been utilized for investigating disease modifiers both by searching for modifiers using genetic screens (21,22), and by candidate approaches (23). Such studies have highlighted the importance of chaperones (22), protein degradation pathways (21), and signaling pathways (24) in polyQ disease process.
Various models have also been developed to mimic Parkinson’s disease and related dementias by expressing α-synuclein in the fly (13). Α-synuclein is the protein that is found in the Lewy body (LB) inclusions in patients with Parkinson’s disease and Lewy body dementia (25). Other studies have focused on Drosophila mutants for parkin, a ubiquitin ligase linked to autosomal recessive juvenile onset parkinsonism (AR-JP) (26–28). The α-synuclein fly model has been shown to recapitulate features of PD including loss of dopaminergic neurons, neuronal LB-like accumulations, and locomotor deficits (13), and a role for chaperones has been uncovered in this model (29). However, there are discrepancies in the literature about the faithfulness of the α-synuclein model as various groups have failed to find any locomotor defects in these flies and also the histopathological features of this model have not proved reproducible between labs (30).
Tauopathies, characterized by the presence of neurofibrillary tangles (NFTs) (for review see (31)), have also been modeled in Drosophila (12). Expression of wild-type or a mutant tau associated with a familial tauopathy leads to adult onset, progressive neurodegeneration, accompanied by reduced longevity, tau accumulation, and increased toxicity of mutant tau (12). However, this model is characterized by lack of the NFTs present in human patients (12). Further studies have used this fly model to investigate the effects of kinases, such as the wingless pathway kinase glycogen synthase kinase (GSK)-3β (in flies, shaggy) (32,33), and also in screens looking for modifiers of tau pathology (34). Interestingly, fibrillar tau pathology was observed when wild-type human tau was co-expressed with shaggy, the main kinase responsible for tau phosphorylation in humans (32). Other studies have utilized the tau model to investigate potential roles of tau, thereby showing a link between human tau overexpression and defects in synaptic transmission in larval neurons (35).
AD is one of the more recent diseases to be modeled in the fruit fly. The rest of this chapter will focus on different Drosophila AD models developed in the last 5 years, how these models have been useful in identifying modifiers of AD, and how they are likely to be used in the future.
2 Making a Model of Alzheimer’s Disease
2.1 Components of AD Pathology
AD was first described by Alois Alzheimer in 1906 as a rare form of presenile dementia; however, it is in fact the most common form of dementia and shows a dramatic increase in incidence with increasing age. It is thought that up to 20 million people worldwide suffer from AD, and the number is projected to double in the next 30 years. The main diagnostic lesions in AD patients are extracellular amyloid plaques and intracellular NFTs. The main component of the senile plaques is a peptide of about 4 kDa, called Aβ peptide, first identified in the plaques of brains of Down’s syndrome patients and subsequently shown to be found in AD brains (36). The Aβ peptide has been shown to be a proteolytic product of the amyloid precursor protein, APP (40). The role of both tau and APP have been investigated in Drosophila with evidence for an axonal transport role for tau and a possible cell–cell adhesion or axonal transport role for APP (10,33,41,42).
The APP holoprotein is a transmembrane protein that becomes metabolized into a variety of peptides, by either the amyloidogenic or the nonamyloidogenic pathway. The catabolism of APP is shown in Fig. 1. In the amyloidogenic pathway, APP is sequentially cleaved by the β- and γ-secretases. The β-secretase activity has been identified to be the beta-site APP cleaving enzyme (BACE) (43,44), whereas the γ-secretase activity appears to arise from a complex of at least four proteins including presenilins 1 and 2, and nicastrin (for review see (45)). The precise subcellular location of proteolytic events is still unclear: cleavage by α-secretase has been shown to occur at the plasma membrane (46), but the β- and γ-secretases are thought to generate Aβ after re-internalization of APP, along the endosomal/lysosomal pathway (47,48). The subcellular location of the β- and γ-cleavage of APP may be significant in disease pathogenesis, as the low pH of the endosomal pathway has been shown to increase Aβ aggregation (49).
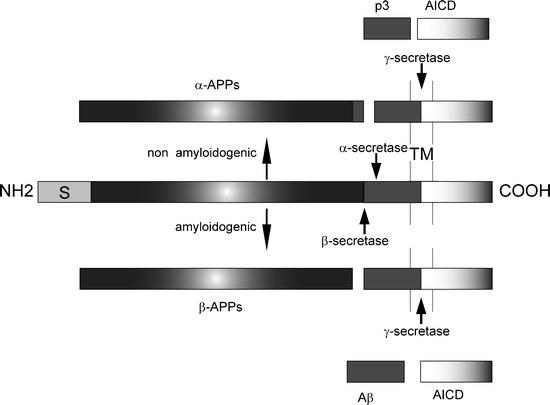
Fig. 1.
Schematic diagram of β-amyloid precursor protein (APP) and its metabolic derivatives.
The APP holoprotein is shown in the middle of the diagram, the signal peptide is marked with S, and the Aβ sequence is shown in dark gray. The Aβ sequence resides partially in the plasma membrane (marked TM). Cleavage of APP by the secretases follows either a nonamyloidogenic pathway or an amyloidogenic pathway (generation of Aβ peptides). Cleavage of APP by α-secretase leads to production of the α-APP soluble fragment and C83, which is further cleaved by γ-secretase to produce p3 and the APP intracellular domain (AICD). Cleavage of APP via β-secretase leads to the production of β-APP soluble fragment and C99, which is further cleaved by γ-secretase to produce Aβ and AICD. Depending on the precise site of cleavage by γ-secretase, the size of Aβ, Aβ peptides of varying lengths are produced, namely 40 or 42 residues long.
2.2 Aβ Is Primary for Neurotoxicity
Various lines of evidence suggest that Aβ42 generation is critical to AD toxicity. First, all FAD mutations are localized in either APP or the presenilin genes (3). Mutations in APP cause FAD (50), either by increasing the aggregation properties of the generated Aβ42 peptide (51), or by increasing the ratio of Ab42 to Ab40 (52). In patients with Down’s syndrome (trisomy 21), FAD is thought to be the result of an extra copy of wild-type APP (APP is on chromosome 21) (53), resulting in increased AD prevalence and reduced age of onset (54). FAD mutations in the components of γ-secretase, presenilins 1 and 2, affect processing of APP and these mutations lead to an increase in Aβ42 production or to an increased ratio of Aβ42 peptide compared to the less aggregatory Aβ40 peptide (55,56). Mutations in the microtubule-associated tau protein have not been shown to cause FAD; however, tau variants are sufficient to cause related neurodegenerative disorders called familial tauopathies (57) that often manifest clinically as frontotemporal dementia.
Despite Aβ42 being the main component of Aβ plaques (58), the number of plaques in a postmortem brain does not correlate well with the severity of AD symptoms in life (60). However, an important step forward in our understanding of the role of Aβ came with the observation that total Aβ levels, which includes soluble forms of the peptide, were found to correlate with disease progression (61). Other evidence points to a role for soluble Aβ42 oligomers in causing toxicity in vivo (66,67), backed by studies where immunization with Aβ oligomer-specific antibodies improves learning and memory in AD transgenic mice (68,69). Taken together, the studies mentioned above point to a role for soluble Aβ aggregates in causing toxicity and being critical for neurodegeneration; furthermore, it appears that Aβ generation is probably sufficient to cause both tau and Aβ pathology.
2.3 Drosophila Models of AD
Two related strategies have been employed in generating models of AD in Drosophila. The first was based on an attempt to closely replicate the APP processing that is found in human patients. Indeed, Drosophila have much of the molecular machinery that is involved in human APP processing, specifically an orthologue of APP, called APPL. Interestingly, the homology to human APP does not stretch to the region that generates Aβ peptides (70). Human APP can rescue APPL mutants (9), and APP overexpression has subsequently uncovered endogenous γ-secretase activity in Drosophila (42). When human β-secretase was cloned (43), the path opened for a complete APP processing Drosophila model. In this model, expression of APP and BACE in the fly eye led to deposition of Aβ plaques and age-dependent neurodegeneration (71). Ubiquitous APP and BACE expression also led to shortened life span and defects in wing vein development (71).
We and others have focused on simpler models, directly expressing various Aβ peptides as secreted peptides in the fly nervous system or in the developing eye (72–74). These models have successfully shown progressive intracellular Aβ accumulation, extracellular Aβ plaque deposition, and neurodegeneration accompanied by olfactory memory defects, reduced longevity, and defective locomotor behavior (72–74). In addition, these models have demonstrated that the phenotypes caused by Aβ production were age- and dosage-dependent, with correlation both between Aβ levels and neurodegeneration (72–74), and also between propensity of Aβ peptide to aggregate into oligomers and the severity of the phenotypes (72,74). Expression of the less aggregatory Aβ40 peptide does not cause neurodegeneration apart from an age-dependent defect in olfactory learning and memory (72). However, a mutation in the Aβ42 peptide (E22G substitution, arctic mutation (51)), which increases its aggregation propensity, leads to more severe, earlier onset of neurodegeneration, locomotor defects, and a markedly reduced survival in comparison with the wild-type Aβ42-arctic peptide (74).
Both types of models have successfully recapitulated many aspects of human AD and each has benefits for studying different aspects of AD neurotoxicity. The APP-based models are good for screening secretase inhibitors (71), and potential modifiers of the effects of secretases on Aβ42 production. The simpler secreted Aβ models are useful for looking at the toxicity of different Aβ peptide variants (75), and for modifiers that affect Aβ metabolism and toxicity. While there appears to be good concordance between the types of phenotypes observed in these two complementary approaches to modeling Aβ toxicity in the fly, there may be some differences in the subcellular localization of the peptide. Specifically, in the normal processing of APP, it is thought that Aβ is generated in an endosomal compartment and may subsequently be released to the extracellular space. In our model system, the peptide enters the secretory pathway from the endoplasmic reticulum (47,48).
3 Characterizing Drosophila AD Model Phenotypes
Some of the phenotypes observed in the Drosophila AD models are analogous to those observed clinically in human patients, such as learning and memory defects, and histopathological phenotypes, such as Aβ plaques and neuronal loss. However, other, easy-to-score phenotypes in the flies are used as surrogate markers for neurodegeneration, such as reduced longevity, locomotor defects, and rough-eye phenotypes.
3.1 Histology
The two main pathologies observed in human AD patients are amyloid plaques and NFTs. In the Drosophila APP and Aβ models, Aβ accumulates intracellularly in the nervous system of the flies, in a manner similar to Aβ accumulation in human AD patient brains. Flies expressing either Aβ peptides or APP/BACE under control of either the neuronal elav c155 -Gal4 or the eye-specific gmr-Gal4 driver are maintained at either normal fly culture temperature (25°C), higher (29°C), or lower (18°C) temperature, in order to accentuate or slow phenotype progression. In order to analyze the progressive Aβ accumulation and neurodegeneration, flies are decapitated, fixed, wax-embedded, and sectioned at different ages and disease stages. For most applications, wax embedding and sectioning are sufficient to obtain material for immunohistochemical analysis (Fig. 2).
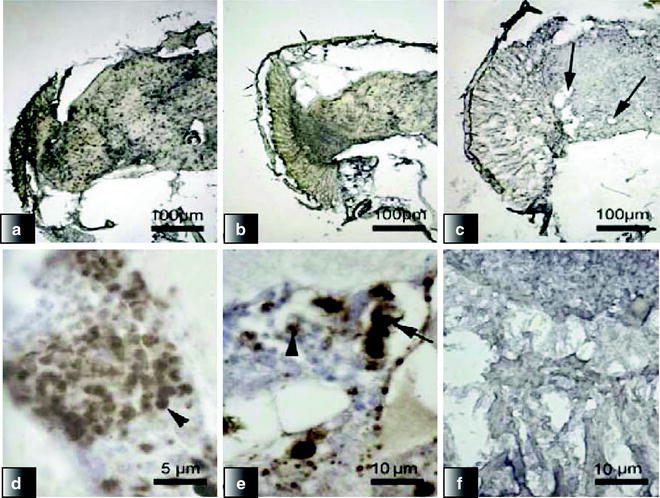
Fig. 2.
Progressive Aβ accumulation, aggregate deposition, and neurodegeneration in the brain and retina of flies expressing Aβ42-arctic under control of Gal4-elavc155.
(a–c) Scanning electron microscopy was used to examine the appearance of the Drosophila eyes (scale bar =100 μm). (d–f) Immunohistochemistry with the 4G8 monoclonal antibody of the retina and brain of flies. On day 0 (a, c), there was widespread, intense intracellular accumulation (arrowhead). Intracellular staining was still present at day 5 (b, e); however, extracellular deposits (arrow) were now visible. By day 9, there was widespread vacuolation (arrows) with concomitant reduction in staining intensity (c, f) (Reproduced from (74)).
Aβ peptides accumulate in an age-dependent manner in the flies’ brains, especially in areas of the brain containing neuronal cell bodies (74). Some deposits are found in aging fly brains, which stain for Aβ; however, they appear not to be mature amyloid deposits as they do not stain with Congo red (74). In other studies, thioflavin S-positive deposits have been identified, and these are comparative to diffuse plaques (71,72). The presence of diffuse nonfibrillar Aβ deposits is supported by positive staining using an oligomer-specific antibody (74,76). Moreover, the Aβ deposition in the flies’ brains is also accompanied by age-dependent neuronal loss (72,74), demonstrating that amyloid plaques are not required for Aβ to cause neuronal toxicity and neurodegeneration.
3.2 Pavlovian Olfactory Learning Assays
The Pavlovian olfactory associative learning abilities of flies have been used to train them to associate an electric shock with a particular odor. Iijima and colleagues used olfactory learning assays to investigate learning and memory in flies expressing secreted Aβ peptides. The loss of this associative learning capacity, or the acceleration of forgetting, may be considered a good analogue of the memory impairment seen in AD patients. They first trained flies by exposure to an electric shock paired with one odor (octanol or methylcyclohexanol) for 60 s and subsequent exposure to a second odor without electroshock for 60 s (72). Immediately after training, learning was measured by allowing flies to choose between the two odors for 120 s. The performance index was calculated by subtracting the number of flies making the incorrect choice from those making the correct one, dividing by the total number of flies, and multiplying by 100. Iijima and colleagues have shown that Pavlovian olfactory associative learning is impaired in flies expressing Aβ peptides that are older than 6 days. Moreover, the degree of impairment is greater in flies expressing Aβ42 than Aβ40 (72).
3.3 Longevity
It is widely accepted that the insulin/insulin-like growth factor (IGF)-like signaling pathway regulates life span across metazoans and much of this evidence has come from studying longevity in invertebrates, such as Drosophila melanogaster (77). However, the precise cause of fly death in general, and in Drosophila AD models in particular, is still unclear, although it is thought that locomotor defects are associated with concomitant feeding deficits. There is a strong correlation between the locomotor ability of the fly and its life span, suggesting that longevity can be used as a surrogate marker for AD as it provides a measure of the health of a group of flies. A main advantage of a longevity assay as a marker is its clear endpoint, as a fly at any time point is either alive or dead. Kaplan–Meyer statistics are used to analyze survival assays of flies and can also take into account censored flies due to escape.
As with many disease-related phenotypes, longevity can be measured at temperatures between 18°C and 29°C, with shorter life spans at higher temperatures. All the groups of flies to be compared in a longevity assay are reared under identical conditions and are placed in groups of ten flies/vial, to a total number of at least 100 flies/genotype. Deaths are scored and flies are transferred to new food medium three times a week. In such assays, flies expressing the nontoxic Aβ40 peptide are used as a control for neuronal peptide expression, and their life span is indistinguishable from that of w1118 control flies (Fig. 3 (72,74)). Flies expressing the toxic Aβ42 peptide have reduced life span compared to elav c155 -Gal4/Aβ40 flies and the effect of Aβ42 expression is dose-dependent, as two copies of the Aβ42 transgene result in a further reduction of life span (Fig. 4 (74)). Expression of the Aβ42-arctic mutant peptide results in a marked reduction in survival compared to expression of wild-type Aβ42 peptide (Fig. 3 (74)).
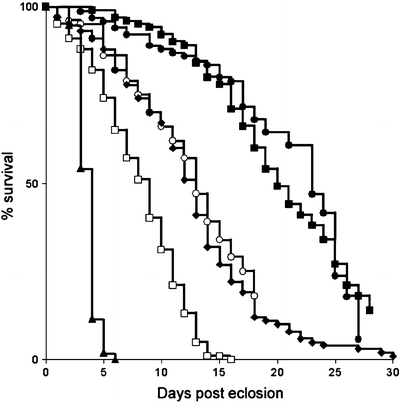
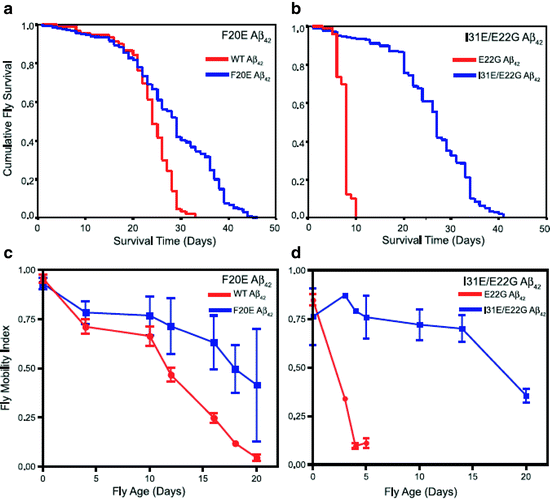




Fig. 3.
Reduced survival of flies expressing Aβ peptides.
The longevity of control w1118 flies (filled squares) was compared with flies expressing one (diamonds and empty circles, two independent insertions) or two copies (empty squares) of the Aβ42 transgene, one copy (triangles) of the Aβ42-arctic or one copy (filled circles) of Aβ40 under the control of the Gal4-elav c155 driver (Reproduced from (74)).
Fig. 4.
Survival and locomotor analysis of various Aβ42 mutants including rational mutations based on computational predictions.
(a) Flies expressing F20E Aβ42 live significantly longer (median survival 29 ± 1 d, n = 400, p < 0.0001) than flies expressing WT Aβ42 (median survival 24 ± 1 d, n = 100). (b) Flies expressing I31E/E22G Aβ42 show a dramatic increase in longevity (median survival = 27 ± 1 d, n = 600, p < 0.0001) compared to flies expressing E22G Aβ42 (median survival = 8 ± 1 d, n = 100). (c) Flies expressing the F20E Aβ42 peptide have significantly improved locomotor ability (p < 0.001, n = 90 observations per line per time point) compared with flies expressing the WT Aβ42 peptide. (d) Flies expressing the I31E/E22G Aβ42 peptide have significantly improved locomotor ability (p < 0.001, n = 90 observations per line per time point) compared to flies expressing E22G Aβ42 (Reproduced from (75)).
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
