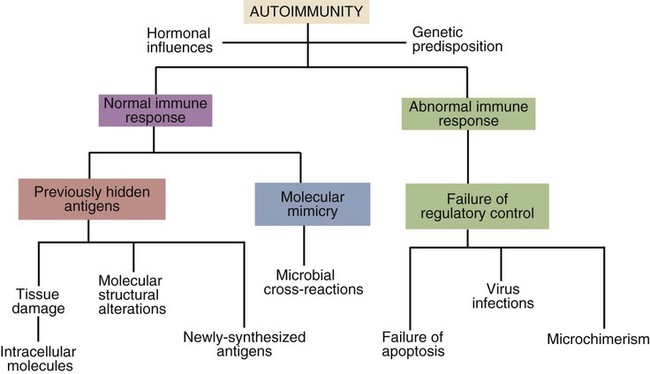
• Autoimmunity is an inescapable consequence of the way in which the adaptive immune system has evolved. • Not all autoimmunity is pathological. Autoimmune responses may be mounted against antigens that develop late in life, against antigens hidden within cells, and against antigens that arise as a result of the development of new molecular configurations. • Most autoimmune diseases result from a failure to ensure that tolerance is maintained against self-antigens. • There may be a strong genetic predisposition to develop autoimmunity. • Some autoimmune diseases are triggered by immune stimulants such as virus infections, vaccination, and some drugs. • The lesions that develop in autoimmune diseases are generated by the mechanisms of hypersensitivity. Autoimmune diseases appear to develop spontaneously, and predisposing causes are rarely obvious. Nevertheless they fall into two major categories: they can result from a normal immune response to an unusual or abnormal antigen, or they can result from an abnormal immune response to a normal antigen (Figure 34-1). The second category is probably the most common. In these cases, the mechanisms that normally prevent the development of self-responsive T and B cells fail. Many different environmental factors and genes contribute to this failure, and the failure may not always be complete. Autoimmune diseases may result from an aberrant response to a single specific antigen; alternatively, they may be due to a general defect in the regulation of B or T cell functions. Potentially harmful, self-reactive lymphocytes are normally destroyed in the thymus by apoptosis triggered through CD95 (Fas) (Chapter 18). Defects in CD95 or its ligand CD154 (CD95L) cause autoimmunity by permitting abnormal T cells to survive and cause disease. This is well demonstrated in the lpr strain of mice. These animals have a mutation that alters the structure of the intracellular domain of CD95 and blocks its functioning. A mutation (called gld) in CD95L has a similar effect. Both lpr and gld mice develop multiple autoimmune lesions accompanied by lymphoproliferation. Some investigators have suggested that mutations in CD95 may contribute to the pathogenesis of lupus in other mammals. The AIRE (autoimmune regulator) gene permits multiple self-antigens to be expressed in thymic epithelial cells. T cells that respond to these self-antigens are destroyed. Thus humans with a defective AIRE gene develop autoimmunity against multiple endocrine organs, the skin, and other tissues. Autoimmunity may result from molecular mimicry, a term used to describe the sharing of epitopes between an infectious agent or parasite and an autoantigen (Figure 34-2). B cells may be triggered by a foreign epitope that cross-reacts with an autoantigen. However, they will only respond to this epitope if they receive T cell help. If nearby Th cells also recognize these microbial epitopes as foreign, they may trigger a response that permits the self-reactive B cells to make autoantibodies. Once a B cell response is triggered in this way, the infectious agent may be removed while the autoimmune response continues—a “hit-and-run” process.
Autoimmunity
General Principles
Induction of Autoimmunity
Abnormal Immune Responses
Failure of Regulatory Control
Infection-Induced Autoimmunity
Molecular Mimicry
< div class='tao-gold-member'>
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Autoimmunity: General Principles

Only gold members can continue reading. Log In or Register to continue
