and David M. Holtzman1
(1)
Department of Neurology, Washington University School of Medicine, St. Louis, MO, Washington, USA
Abstract
The PDAPP mouse has been a very useful model for studying mechanisms underlying amyloid-β (Aβ) metabolism, aggregation, and deposition, and for aiding in the development of new diagnostic and therapeutic approaches for Alzheimer’s disease (AD). One of the initial difficulties in producing a mouse model that was useful to study AD was creating an easily manipulated small animal model that demonstrated some of the key in vivo neuropathological hallmarks of the disease. The PDAPP model was the first transgenic mouse to overexpress the human amyloid precursor protein (hAPP) that successfully recapitulated several neuropathological features characteristic of AD. These include Aβ deposition in both diffuse and neuritic plaques, cerebral amyloid angiopathy, astrocytosis, microgliosis, hippocampal atrophy, synaptic alterations, and behavioral deficits. Many of the histological, biochemical, and structural alterations present in the PDAPP mouse closely resemble the changes found in the brain of AD patients, especially the temporal and spatial-specific deposition of Aβ in the brain. The animals, however, did not develop tauopathy or significant neuronal cell death. Because of these properties, the PDAPP mouse has proven to be an attractive model to study the disease process underlying aspects of AD that are related to Aβ aggregation and its consequences. The PDAPP mouse has been extensively used to study the effect of genetic factors that modify AD, as well as Aβ-binding proteins and their effect on Aβ deposition. It has also been widely used to characterize the potential use of active and passive immunization in both the diagnosis and treatment of AD.
Key words
PDAPP mouse modelAmyloid beta (Aβ)Platelet-derived growth factor (PDGF)-βhuman amyloid precursor protein (hAPP)Amyloid plaqueCerebral amyloid angiopathy (CAA)Neuritic dystrophyApolipoprotein EClusterinImmunization1 Introduction
Mouse models have proven to be valuable tools for both elucidating the pathophysiology of various disease states and for studying new treatments. The study of Alzheimer’s disease (AD) in particular has benefited in recent years from the development of multiple mouse models that exhibit various characteristics of the disease. An initial challenge in studying AD in vivo was creating a mouse model that successfully displayed the neuropathological hallmarks of AD, including extracellular amyloid-β (Aβ) deposition, inflammation, neuritic dystrophy, intracellular neurofibrillary tangles, and neuronal as well as synaptic loss. Investigators began to develop such a model by overexpressing the human amyloid precursor protein (hAPP) with or without mutations that result in early-onset forms of familial, autosomal dominant AD. Early attempts at developing hAPP transgenic mice did not achieve their main objective as the brains of these animals had low levels of Aβ deposition or no Aβ accumulation at all (1–7). However, in 1995 a team of scientists from Athena Neurosciences greatly facilitated the study of AD in mice by developing the PDAPP mouse model (8). This was the first APP-based transgenic mouse to display many of the pathological features of AD, including Aβ deposition in both diffuse and neuritic plaques, astrocytosis, microgliosis, hippocampal atrophy, synaptic alterations, and behavioral deficits. This chapter will summarize the design and characterization of the PDAPP mouse, and also highlight how this model has been used to understand the mechanisms underlying the pathogenesis of AD, as well as to develop better diagnostics and treatments.
2 Design and Biochemical Characterization of the PDAPP Model
Like many other AD mouse models, the generation of the PDAPP mouse relied on previous genetic studies that revealed that multiple mutations in the hAPP gene could cause familial, autosomal dominant forms of AD. The genetic construct used in creating the PDAPP mouse consists of an hAPP minigene with a single-point mutation that contains a phenylalanine substituted for valine at residue 717 (V717F, also known as the Indiana mutation (9)), expressed under the control of the platelet-derived growth factor (PDGF)-β promoter (8). The PDGF-β promoter was chosen because it preferably targets expression of the transgene to neurons (10). The V717F mutation has been shown to increase the relative production of the more amyloidogenic Aβ42 in relation to other shorter Aβ species such as Aβ40, which most likely plays a significant role in the propensity for Aβ to aggregate in the brain of PDAPP mice (11). To allow for the alternative splicing of exons 7 and 8 that is known to occur with human and mouse APP, the hAPP (V717F) construct also contains introns 6–8 (8, 12). Analysis of protein expression showed that the hAPP protein in PDAPP mice is expressed at a level that is approximately 10-fold higher than both the levels of APP found in the human brain and the amount of endogenous mouse APP in the PDAPP brain (8). Studies have also shown that the hAPP protein is expressed predominantly in the brain of PDAPP mice, demonstrating that most of the human Aβ produced is of CNS origin (13).
A very useful feature of the PDAPP model lies in the fact that the deposition of Aβ occurs in an age-dependent and region-specific manner that is similar to what is found in the human AD brain. Biochemical analysis of Aβ levels in the PDAPP mouse brain demonstrated a progressive increase in the total Aβ levels found in both the hippocampus and cortex beginning around 6–8 months of age in heterozygous mice, with greater levels occurring in the hippocampus (14). This accumulation correlated with the steady increase in immunoreactive Aβ deposition that occurred as the mice aged (see the next section). The accumulation of Aβ42 that occurred dramatically increased with age, likely as a result of the increased levels of Aβ42 aggregating in plaques (14, 15). The mechanism underlying the age-dependent nature of cerebral Aβ deposition in these mice is unlikely to be due to increased production of Aβ with age, as experiments proved the processing of hAPP to Aβ to remain relatively stable as the PDAPP mice age (14). Just as age seems to play a prominent role in Aβ build-up, the region of the PDAPP brain is also important in determining where Aβ deposits. Specific regions of the cortex and hippocampus exhibited significantly greater Aβ accumulation than the thalamus and cerebellum (14, 16, 17). Experiments have shown that even though the level of hAPP is greater in both the thalamus and cerebellum of PDAPP homozygous mice in comparison to the level of hAPP in the cortex of heterozygous mice, the heterozygous cortex developed greater levels of Aβ accumulation (14). Therefore, Aβ deposition occurs in specific regions of the brain in a manner that in some cases is not completely predicted by hAPP levels.
The distribution and metabolism of Aβ in the PDAPP mouse has been closely analyzed in specific compartments of the CNS and in the plasma. Both cerebral spinal fluid (CSF) and plasma contained measurable levels of soluble Aβ that exist in a close equilibrium in mice without plaque deposition (18). However, once plaques began to accumulate, CSF Aβ levels, particularly Aβ42, decreased and the correlation between plasma and CSF Aβ dissipated, suggesting that Aβ deposition alters the equilibrium between these compartments by increasing the propensity of Aβ to remain in the CNS, with Aβ aggregates acting as a local “sink” for soluble Aβ (15, 18). Subsequent data were obtained using in vivo microdialysis that showed that plaque deposition is associated with altered soluble Aβ homeostasis in the brain interstitial fluid (ISF). The half-life of ISF Aβ was about twofold longer in 12-month-old PDAPP mice with plaques than in 3-month-old mice without plaques (15). Finally, Aβ accumulation also occurred in the cerebral vessels as PDAPP mice aged, resulting in cerebral amyloid angiopathy (CAA) (19, 20). Interestingly, the deposition of Aβ in vessels resulted in a significant decrease in the Aβ42/Aβ40 ratio in the Aβ aggregates located within the vessel wall, which is similar to what is found in human brains with CAA (20). Therefore, unlike in the brain parenchyma where Aβ42 is the most abundant species present in plaques, Aβ40 appears to be the primary depositing species of Aβ in vessels.
3 Histopathology of the PDAPP Mouse Brain
Many of the pathologic changes that occur in the PDAPP mouse brain resemble the histological hallmarks that occur in the human AD brain. The progressive development of both neuritic and diffuse Aβ deposits is the primary histological feature in the PDAPP brain, along with secondary changes in parenchymal and synaptic structure. On average, Aβ deposition visible by immunostaining starts to occur around 6–8 months of age and increases progressively as the animals age (14, 16, 21). However, Aβ immunoreactivity has been detected in some homozygous animals at ages as young as 3–4 months old (21). Furthermore, multiple studies have revealed that the age in which visible Aβ deposition begins varies significantly, as heterozygous mice up to 12 months of age have shown no Aβ accumulation (13, 18, 22, 23). Aβ deposits tend to first occur in the cingulate cortex and then soon after or concurrently appear in the molecular layer of the dentate gyrus, the CA1 region of the hippocampus, the entorhinal cortex, and the corpus callosum (8, 16). Very little deposition has been found in the cerebellum and thalamus (8, 17). Multiple studies have shown that the greatest Aβ load occurs in the molecular layer of the dentate gyrus and the lateral entorhinal cortex, suggesting that the lateral perforant pathway in these animals is particularly subject to Aβ deposition (16, 17, 24). Approximately 10% of the Aβ deposits covering the brain stained with thioflavine-S, which is synonymous with Aβ aggregation into amyloid fibrils or fibrillar plaques that have a β-pleated sheet structure (8, 25). The morphology of these plaques varied from irregular immunoreactive deposits that lack a central amyloid core to spherical complex structures with a dense amyloid core (see Fig. 1) (8, 25, 26). Also, fibrillar (thioflavine-S-positive) plaque accumulation associated with CAA occurred in the wall of cerebral arterioles starting around 15 months of age (19, 20, 27, 28). The progression of CAA in the PDAPP brain was associated with the development of microhemorrhages as the mice age (19), presumably due to CAA-associated vessel damage.
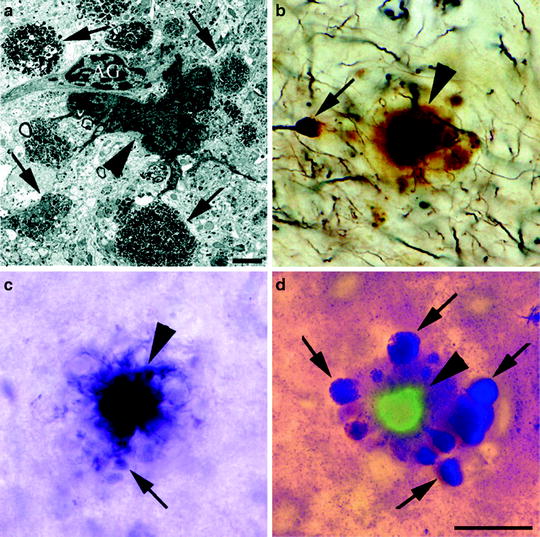
Fig. 1.
Neuritic plaque morphology present in PDAPP mice.
(a) Appearance of an amyloid plaque by transmission electron microscopy highlights the extracellular amyloid deposit (arrowhead), dystrophic neurites (arrows), and astroglial cells (AG) associated with the plaque. Plaques stained by Bielschowsky (b), Gallyas (c), and de Olmos (d) silver staining methods display the dense amyloid core (arrowheads) and dystrophic neurites (arrows). The section in (d) is also stained with thioflavine-S to emphasize fibrillar Aβ present in the amyloid core (Reprinted from (60). Copyright 2003 with permission of Wiley-Liss, Inc., a subsidiary of John Wiley & Sons, Inc.).
Dystrophic neurites and changes in the neuropil structure are closely associated with plaque deposition in the PDAPP brain (8, 26). An extensive comparison of both human AD brains and the PDAPP brain has shown structural similarities between the dystrophic neurites in both cases, including the presence in dystrophic neurites of dense multilaminar bodies, dense-core and clear synaptic vesicles, and neurofilament accumulation (26). Both astrogliosis and elevated levels of activated microglia were prominent in the regions with extensive Aβ immunoreactivity (8, 16, 26, 29). Interestingly, as in the human brain, gliosis was mostly centered around fibrillar plaques, not diffuse plaques (26). Finally, it should be noted that though extracellular Aβ accumulation is extensive in the PDAPP mice, intracellular tau pathology is minimal. Staining for phosphorylated tau and phosphorylated neurofilaments showed a very slight increase in a minority of dystrophic neurites as the PDAPP mice age (26, 30). Most dystrophic neurites in PDAPP mice do not contain abnormal tau immunoreactivity (26, 30). Paired helical filaments characteristic of neurofibrillary tangles have not been observed in the PDAPP brain (8, 26, 30), nor in other APP transgenic mice that develop Aβ deposits unless the human tau gene is also introduced into mice (31).
Pathological analysis of the PDAPP brain also revealed structural alterations in the neuronal parenchyma and synaptic network both before and after Aβ deposition occurs. Multiple studies have reported hippocampal atrophy present in the PDAPP mice as early as 3 months of age, thus before plaque accumulation occurs (21, 32, 33). Whether brain changes such as hippocampal atrophy, which are present in young PDAPP prior to Aβ aggregation, are due to overproduction of APP or APP fragments, soluble forms of Aβ, or another cause has not been clearly demonstrated. Hippocampal atrophy was particularly prominent in the dentate gyrus (33). High-resolution magnetic resonance spectroscopy illustrated that there is a 12% reduction in hippocampal volume in 100-day-old PDAPP mice in comparison to wild-type mice that remained constant as the mice age (33). However, hippocampal atrophy is not likely due to a decrease in neurons, as another study has shown that there is no neuronal loss in the entorhinal cortex, cingulate cortex, or CA1 region of the hippocampus (16). Whether or not synaptic density is altered in the PDAPP brain is still debatable, as a few studies looking at synaptophysin immunoreactivity have described either a decrease in synapses (8, 21), or no change (16). Another report showed that the level of cholinergic nerve terminals in the cerebral cortex was decreased in young PDAPP mice, yet analysis of these same mice did not show a loss of cholinergic cell bodies (34). Therefore, the possibility still remains that Aβ accumulation and deposition in the PDAPP brain either directly or indirectly causes modification of neuronal networks that may lead to some of the behavioral changes and synaptic dysfunction that exist in these mice, particularly as they age (see below).
4 Behavioral Deficits and Synaptic Dysfunction in the PDAPP Model
Behavioral analysis of the PDAPP mouse has revealed the presence of multiple age-dependent and independent deficits that exist in both learning and memory paradigms. Initial observations of these mice demonstrated that both heterozygous and homozygous animals exhibit significant impairment in a radial-arm maze task. A substantial increase in the number of reference and working memory errors occurred in 3, 6, and 9- to 10-month-old PDAPP mice in comparison to wild-type mice (35). In an object recognition task, homozygous 6-month-old mice, and both heterozygous and homozygous 9- to 10-month-old mice showed significant impairments indicative of an age-dependent decline in recognition performance (35). Combining both behavioral data and the pathological characterization of these mice, the authors of this study concluded that the decline in object recognition capability correlates with the progression of Aβ deposition, while spatial reference and working memory deficits are independent of Aβ accumulation and may relate to hippocampal atrophy and synaptic loss due to other reasons such as APP overexpression (21). Another study using a modification of the water maze in which the mice must learn successive platform locations to test working memory demonstrated that PDAPP mice have an age-independent deficit in learning the location of the first platform, and also an age-dependent deficit in learning later platform locations (36). This age-related decline in learning ability correlated with amyloid plaque load and was apparent using both a cross-sectional study of mice of various ages as well as using a longitudinal approach in which the same PDAPP mouse was tested repeatedly with age (36). Using a similar water maze test, another research team used swim distance as the measure of learning performance and showed that both young and old mice were significantly impaired in spatial learning (37). Importantly, there were both age-dependent and independent learning abnormalities. The age-dependent effects are probably related, at least in part, to Aβ accumulation. Other tests show that alterations occur in the PDAPP mice in behavioral patterns not related to cognition, including changes in core body temperature, sleep-wake states, and fear and eye-blink conditioning (32, 38, 39). Therefore, the behavioral phenotype present in PDAPP mice is complex with possibly a developmental defect that leads to learning and other impairments. This defect may be due to overproduction of APP or APP fragments.
A small amount of information has been published demonstrating changes in synaptic function in the PDAPP brain. Electrophysiological studies have shown that different changes in synaptic activity occur in young versus old PDAPP mice (40–42). Young mice (4- to 5 months old) had a greater level of paired-pulse facilitation (PPF) and long-term potentiation (LTP) that decayed at a much more rapid rate than in wild-type mice (40). In older mice (27- to 29 months old), there was also a reduction in PPF, but the difference in LTP in comparison to wild-type control mice was not as robust as in the younger mice. The synaptic response in older mice was also greatly reduced (40). Another study analyzed the extent of cholinergic dysfunction in PDAPP mice and found that basal acetylcholine (ACh) levels were lower in PDAPP mice (from 2 to 6 months of age) compared to nontransgenic mice (43). Also, significant alterations were found in the levels of ACh released following both pharmacological and environmental stimuli (43). Therefore, it appears that in the PDAPP mice both synaptic activity and the extent of neurotransmitter release are affected, though the relationship between these changes and behavior is not yet clear.
5 The PDAPP Model and the Study of Aβ-Binding Proteins
The PDAPP mouse model has played an important role in studying mechanisms underlying AD pathogenesis. In particular, the model has been utilized to examine the in vivo relationship between Aβ and its binding proteins, including apolipoprotein E (apoE), clusterin (also known as apolipoprotein J), and α-1-antichymotrypsin. In humans, apoE genotype is the most important genetic risk factor for AD (44, 45). PDAPP mice were crossbred with apoE-/- mice. PDAPP mice deficient in murine apoE had a dramatic dose-dependent reduction in the extent of Aβ immunoreactivity in both the hippocampus and cortex (46–49), with almost no Aβ deposition in the cortex of apoE-/- mice up to 22 months of age (see Fig. 2) (47). Interestingly, the brains of PDAPP/apoE-/- mice also showed a virtual complete absence of thioflavine-S-positive plaques up to 22 months of age (46, 47). The anatomical distribution of Aβ that did deposit in the hippocampus of the PDAPP/apoE-/- mice was different from PDAPP/apoE+/+ mice, with most deposition occurring in the hilus of the dentate gyrus and very little in the molecular layer (48, 49). Also, very few neuritic plaques were found in the hippocampus of the PDAPP/apoE-/- mice (49), and almost no CAA or microhemorrhages were present in these mice through 24 months of age (19). Therefore, these studies demonstrate that murine apoE is necessary for the deposition of fibrillar Aβ that is present in both neuritic plaques and CAA in PDAPP mice.
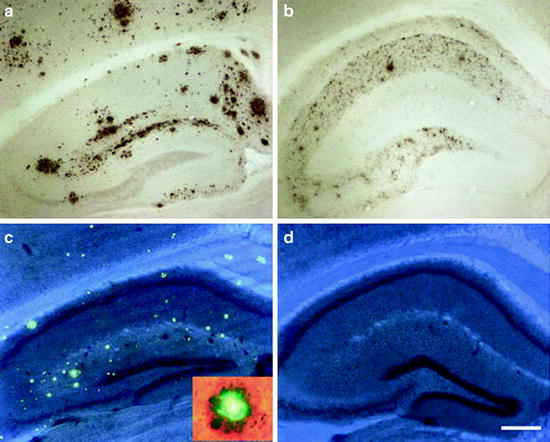
Fig. 2.
The role of mouse apoE on Aβ and amyloid deposition.
Brains from PDAPP mice that express murine apoE have significantly greater levels of Aβ deposition (a) and thioflavine-S stained plaques (c) in both the hippocampus and cortex in comparison to mice lacking apoE (b,d). PDAPP, apoE-/- mice are virtually deficient in thioflavine-S positive deposits (d). The inset in (c) shows a thioflavine-S plaque surrounded by dystrophic neurites (Reprinted with permission from Macmillan Publishers Ltd., Molecular Psychiatry, (61), 2002).
The PDAPP mouse has also been crossed to clusterin-/- mice. Brains from PDAPP mice lacking clusterin did not show a change in the levels of Aβ deposition or in the age at which deposits occur (50). However, these brains do have a marked decrease in fibrillar plaque levels and neuritic dystrophy (50, 51). Interestingly, PDAPP mice deficient in both apoE and clusterin (apoE-/-/clusterin-/-) have earlier Aβ deposition and increased levels of both Aβ and amyloid in comparison to apoE-/- and clusterin-/- mice (51). The elimination half-life of ISF Aβ measured by in vivo microdialysis is also significantly decreased in the PDAPP/apoE-/-/clusterin-/- mice and in PDAPP/apoE-/- mice (51). These findings suggest that apoE and clusterin are able to cooperatively regulate Aβ deposition, and that apoE plays a role in regulating the metabolism of extracellular Aβ. Finally, PDAPP mice crossed with α-1-antichymotrypsin transgenic mice had a greater plaque load and density than normal PDAPP mice, along with increased Aβ levels in the brain (52). Alpha-1-antichymotrypsin, therefore, may play a role in promoting Aβ deposition and fibrillogenesis in the PDAPP mice.
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


