and Peter Paul De Deyn1, 2
(1)
Laboratory of Neurochemistry and Behaviour, Institute Born-Bunge, University of Antwerp, Wilrijk, Belgium
(2)
Department of Neurology, Middelheim Hospital, ZNA, Antwerp, Belgium
Abstract
Early-onset familial and late-onset dementia of the Alzheimer-type account for the major proportion of cases of dementia and of neurodegenerative diseases in general. The number of affected individuals is likely to grow in the decades to come due to demographic changes and rising life expectancy. Considerable research efforts are directed at the development and validation of animal models of AD, aiming to further unravel the underlying degenerative processes and searching for therapeutic strategies to alleviate or prevent this devastating condition. These models have contributed to a growing understanding of the molecular pathways involved in disease development and progression. The successful use of animal models in drug discovery relies on both the development of valid disease models and the availability of adequate testing paradigms for the evaluation of the effects of different therapeutic approaches aiming at symptomatic treatment of memory impairment or behavioral alterations, or at disease-modification and/or neuroprotection.
Key words:
Drug discovery pipelineanimal modelsvalidationdementiafenotype1 Drug Discovery Pipeline
The general drug discovery pipeline is depicted in Fig. 1. Animal models – or laboratory animals in general – play an essential role in various stages of the drug discovery pipeline. In target discovery and validation, molecular targets pivotal to the disease process are studied. A target may be a receptor, proteins (enzymes), DNA, RNA, or ribosomes. In addition, the “drugability” of a target, which may be influenced by cellular location, development resistance, transport mechanisms, side effects, and toxicity, is assessed. Assay development encompasses the development of valid model systems, both in vitro and in vivo. This stage has known a major boost with the development of transgenesis and gene targeting techniques, but is, nevertheless, still the bottleneck of the drug discovery pipeline, since all models are partial and for some human diseases (e.g., hepatitis C infection) adequate models are still completely lacking. When converging hits-to-leads, animals are used to assess the potency (ED50) and dose-response curve of a compound. The mechanism of drug action is studiedin vitro, on whole-cell bases systems and in animal models. During lead optimization, different species are used to study pharmacokinetics, pharmacodynamics, absorption, distribution, metabolism, and excretion, as well as to optimize formulation and delivery of the lead. All nonclinical data are compiled during the development stage. If required, some additional preclinical studies may be initiated before proceeding to the clinical phases.
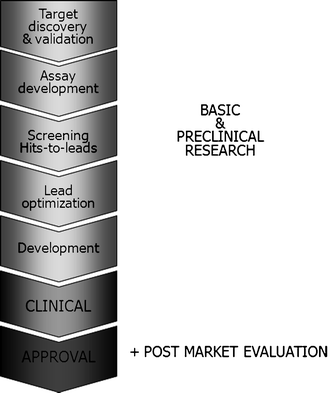
Fig. 1.
Drug discovery pipeline. The drug discovery pipeline in general consists of three major stages: basic and preclinical research, clinical studies, and the (post) approval phase. Animal models – or laboratory animals in general – play essential roles in the drug discovery pipeline. See text for more detailed information on the use of animal models during the different preclinical stages.
2 Validation of Animal Models
Valid animal models are indispensable to the drug discovery and development pipeline. In particular, animal models have a key role to play in target discovery and validation, which require proof that a molecular target is pivotal to the disease process, and that modulation of that target has therapeutic effect. The development of relevant transgenic (knock-out/in) or chemical knock-out models are vital to this stage. Additionally, the use of valid animal models for a specific human condition allows appraisal of the preclinical efficacy of candidate drugs.
The quality and utility of any animal model should be assessed through rigorous validation. A valid model resembles the human condition in etiology, pathophysiology, symptomatology, and response to therapeutic interventions. As a result, homologous models with etiology and symptomatology identical to the human condition exhibit the highest level of validity. In isomorphic models, symptomatology is not provoked by the same factors as in the human condition, leading to reduced validity (2).
The conclusions drawn from animal models largely depend on the validity of the model in representing the human condition. The following perspectives are commonly used in the characterization of a model’s validity. Face validity refers to the resemblance between model and situation or process being modeled. It refers to the phenomenological similarity between the model and the human condition. Similarity of symptoms is most commonly used to assess face validity.
Predictive validity is an empirical form of validity that represents the extent to which the performance of the animal model in a test predicts the performance in the condition being modeled. This level of validation, therefore, necessitates parallel development of clinical measures for meaningful comparisons between model and man. In a more narrow sense, this term is sometimes used to indicate pharmacological isomorphism, i.e. the model’s ability to identify compounds with potential therapeutic effects in the human condition. Construct validity refers to the theoretical clarification of what a test measures or a model is supposed to mimic. Because a given condition may manifest itself in different ways in different species, the behavior used in the animal model may not match that of humans, yet the test or model may still be valid. Construct validation is useful in the incessant process of further developing and refining an animal model. Etiological validity is closely related to construct validity, and refers to identical etiologies of phenomena in the model and the human condition. Models with high etiological validity are most valuable in drug development and discovery.
The more levels of validity a model satisfies, the greater its value, utility, and relevance to the human condition. A “perfect” model would account for etiology, symptomatology, treatment, and physiological basis. Animal models in general do not meet all of these criteria.
3 Validation of Animal Models of Dementia
The key etiological and symptomatic features of AD that would ideally be replicated in an animal model of the disease are outlined in the following paragraphs. In reality, most animal models are partial models, focusing only on restricted aspects of a disease, and modeling the complete condition is often not pursued.
As the prototype of cortical dementias, AD presents with prominent cognitive deficits. Initially, patients display limited forgetfulness with disruption of memory imprinting, evolving to short-term memory disruption, and with disease progression, long-term memory deficits. Most cortical dementias are characterized by the early-stage development of anomia progressing towards aphasia with perseverations and paraphasia. In addition, patients exhibit agnosia, including prosopagnosia or face blindness (representing a major burden for family members), as well as temporal and spatial disorientation. In a more advanced stage, apraxia results in executive dysfunctioning and, hence, the increasing helplessness of the AD patient (3). Besides cognitive deterioration, patients display noncognitive symptoms, such as anxiety, depression, aggression, agitation, hallucinations, circadian rhythm disturbances, and wandering. In contrast with cognitive symptomatology, these noncognitive symptoms – commonly referred to as behavioral and psychological signs and symptoms of dementia (BPSD) – do not exhibit a progressive course. The impact of BPSD is emphasized by the fact that they increase patient suffering, impose tremendous strain on family members and caregivers (often motivating institutionalization), and increase the financial burden on the family and society (3,4).
The histopathological hallmarks of AD brain are extracellular amyloid plaques and intracellular neurofibrillary tangles (NFT), accompanied by decreased synaptic density, which eventually leads to widespread neurodegeneration, loss of synapses, and failure of neurotransmitter pathways, particularly those of the cholinergic system. Clinical, epidemiological, and biochemical research over roughly the past 2 decades has significantly increased our knowledge of the pathological and molecular cascades underlying AD, uncovering potential biomarkers for disease progression or early diagnosis and revealing new drug targets. For a therapeutic intervention to slow down or halt disease progression – that is, to be disease-modifying – it must interfere with these central pathophysiological pathways. The following key pathways are being extensively scrutinized in AD rodent models.
3.1 The Amyloid Hypothesis
The hypothesis that cerebral accumulation of amyloid β (Aβ) peptides in amyloid plaques is the primary culprit driving AD pathogenesis has dominated research for 2 decades. Further disease processes are proposed to result from an imbalance between Aβ production and clearance. Cleavage of amyloid precursor protein (APP) by α-secretase precludes release of Aβ peptides (nonamyloidogenic pathway), whereas the combined effect of β- and γ-secretase cleavage releases Aβ peptides of various lengths (amyloidogenic pathway) (Fig. 2). Some early-onset AD cases showed an autosomal dominant inheritance pattern, leading to the discovery of several major disease genes associated with amyloid processing (5), including mutations in APP, and in presenilin-1 (PSEN-1) and presenilin-2 (PSEN-2), proteins that form the catalytic unit of the γ-secretase protein complex (6). Another recent hypothesis attributes synaptic failure to the presence of soluble Aβ oligomers or amyloid-derived diffusible ligands (commonly abbreviated as ADDLs, pronounced “addles”), which can rapidly block long-term potentiation, and hence cause memory failure (7).
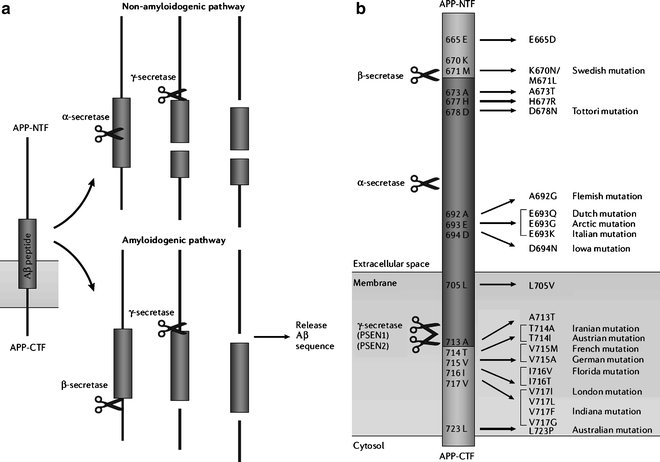
Fig. 2.
APP processing and APP mutations associated with early-onset Alzheimer’s disease. (a) Amyloid precursor protein (APP) processing involves proteolytic cleavage by several secretases. The nonamyloidogenic pathway is initiated by α-secretase cleavage occurring in the middle of the amyloid β (Aβ) sequence, and results in the release of several soluble APP fragments. The amyloidogenic pathway releases Aβ peptides through subsequent cleavage by β and γ-secretase. (b) Part of the APP amino acid sequence with indication of mutations associated with early-onset Alzheimer’s disease. Most mutations are clustered in the close vicinity of secretase cleavage sites, thereby influencing APP processing, and are named after the localization of the first family in which a specific mutation was demonstrated. From (1). Legend: A, alanine; Aβ, amyloid β; APP-NTF, N-terminal fragment of the amyloid precursor protein; APP-CTF, C-terminal fragment of the amyloid precursor protein; D, aspartic acid; E, glutamic acid; F, phenylalanine; G, glycine; H, histidine; I, isoleucine; K, lysine; L, leucine; M, methionine; N, asparagine; P, proline; PSEN, presenilin; Q, glutamine; R, arginine; S, serine; T, threonine; V, valine; Y, tyrosine.
3.2 Tau-Phosphorylation, Paired Helical Filaments, and NFT
A second hallmark of the AD brain is the presence of intracellular NFT, resulting from the hyperphosphorylation and aggregation of CNS tau proteins, a group of microtubule-associated proteins that contribute to the assembly and stabilization of microtubules. Tau function is determined by the degree of phosphorylation, which is regulated by a balance between several protein kinases and phosphatases, with hyperphosphorylation preceding the formation of paired helical filaments and therefore NFT.
3.3 Oxidative Stress
Oxidative damage to different classes of biological macromolecules (sugars, lipids, proteins, and DNA) is a hallmark of both normal aging and most neurodegenerative diseases (8). Multiple lines of evidence demonstrate that oxidative stress is an early event in AD, and therefore may play a key pathogenic role. Oxidative stress may be mitigated through the use of antioxidants, and in the event of increased oxidative stress, neurons themselves upregulate antioxidant defense systems. Evidence even indicates that in the initial phase of AD, Aβ deposition and hyperphosphorylated tau represent a primary line of defense against oxidative stress. With disease progression, the brain is challenged with a profound redox imbalance, resulting in a transformation of both substances into pro-oxidants (9).
3.4 Inflammation
Many neuroinflammatory mediators are upregulated in affected areas of the AD brain, including prostaglandins, complement components, anaphylatoxins, cytokines, chemokines, proteases, protease inhibitors, adhesion molecules, and free radicals (10). Epidemiologic data suggests that prolonged use of nonsteroidal anti-inflammatory drugs (NSAIDs), which target cyclooxygenase (COX), a key mediator of the inflammatory cascade, for conditions like arthritis entails a reduced risk and delayed onset of AD. Initially, the effect of NSAID use in AD was attributed to a reduction of inflammation, but several clinical trials testing NSAIDs in AD patients yielded negative results (10 – 12) and in 2001; however, it was reported that a subset of NSAIDs reduced Aβ1–42 production in cultured cells and the mouse brain (13), effects that are independent of COX inhibition. However, the initial rationale for the use of NSAIDs in AD – that is classical anti-inflammatory mechanisms – should not be completely abandoned.
3.5 Lipid Metabolism and Apolipoprotein E
Several lines of evidence support a strong link between lipid/cholesterol metabolism and dementia. Apolipoprotein E (ApoE) is a plasma and cerebrospinal fluid protein that serves as a ligand for low-density lipoprotein receptors and is involved in the transport of cholesterol and other lipids among various cells of the body. The ApoE ε4 allele in particular is genetically associated with both vascular and Alzheimer dementia (14). Increased dietary intake of cholesterol promoted amyloid deposition in rabbit brain (15), whereas statins were able to reduce Aβ peptide levels both in vitro and in vivo, in guinea pig cerebrospinal fluid (16).
3.6 Neuronal and Synaptic Loss
The mechanisms underlying AD-related neuronal loss are not clear-cut. Apoptosis, triggered by Aβ, activation of glutamate receptors, oxidative stress, DNA damage, and elevation of intracellular calcium levels may occur, as well as other forms of programmed cell death or necrosis. Synaptic dysfunction or loss contributes to clinical symptomatology through disruption of neuronal communication and occurs early in the disease process, long before neuronal and synaptic loss, the latter currently representing the best pathological correlate of cognitive decline (17).
4 Preclinical Drug Evaluation in AD Models
In general, animal models of human disease can be classified into spontaneous, induced, negative, and orphan models, although the two latter types are not relevant when considering dementia models. Although spontaneous models are presumed to develop their condition without experimental manipulation, selective breeding is often necessary to establish and maintain the mutant line. Induced models display conditions as a result of artificial manipulation. Particularly for psychiatric and neurological conditions, few spontaneous models exist, and experimentally induced pathology is often necessary (3). Although models based on many different species, including primates and other mammals, as well as invertebrates, have contributed to the wider field of AD research, rodent models are predominant in drug discovery. The different species and types of models available for dementia-related research will be dealt with in a different chapter of this volume.
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


