, Masashi Kitazawa1 and Frank LaFerla1
(1)
Department of Neurobiology and Behavior Institute for Brain Aging and Dementia, University of California, Irvine, CA, USA
Abstract
Alzheimer’s disease (AD) is a devastating disease, and the most common form of dementia to afflict the elderly population. The disease causes a slow but progressive neurodegeneration, leading to memory impairments and dysfunction in other cognitive domains. The molecular mechanism of disease development and progression has not yet been fully established, nor have any cures or effective, long-lasting treatments been developed. Various transgenic mouse models of AD have proven to be invaluable tools for elucidating disease mechanisms and for providing a platform to evaluate therapeutic strategies. In this chapter, we discuss findings from the 3xTg-AD mouse model, which develops both plaque and tangle pathologies, the two major pathological hallmarks of AD. Studies using the 3xTg-AD mice have revealed a strong interaction between amyloid-beta (Aβ) and tau, which synergistically drive the pathogenesis in the brain.
Key words
Alzheimer diseaseAmyloid-betaTau3xTg-AD miceAnimal models1 Introduction
Alzheimer’s disease (AD) is the leading cause of dementia in the elderly population, afflicting nearly 24 million people worldwide (1). AD patients suffer through a progressive cognitive decline, characterized by the devastating loss of semantic and episodic memory, impairments in judgment, spatial orientation, and language. The disease process is believed to be triggered by the accumulation of the amyloid-beta (Aβ) peptide, which lies upstream of the other pathological changes. AD brains are marked by two pathological hallmarks, amyloid plaques (comprised of Aβ) and neurofibrillary tangles (NFTs), which are made mostly of hyperphosphorylated tau protein (2, 3). Other pathological changes occur as well, including the occurrence of dystrophic neurites, synapse loss, and neuron loss. Currently, no effective treatment exists that either halts or slows the progression of the disease. Thus, there is a great need to more fully understand the disease and to develop and test potential therapies for clearing the pathology and restoring cognition. In the past 2 decades, research has advanced significantly due to the identification of genetic components of AD, and the subsequent development of transgenic mouse models that mimic AD pathology and cognitive decline. These transgenic mice have helped expand our understanding of AD pathogenesis and have provided insights into possible therapies.
2 Transgenic Mice and AD
Genetic analysis of rare, autosomal dominant, early-onset AD identified Aβ as the main culprit behind the pathogenesis of the disease (4). Mutations in the presenilins (PS-1, PS-2) or the amyloid precursor protein (APP) lead to either increased production of total Aβ, the longer more amyloidogenic Aβ1-42, or increase the aggregation of Aβ (5). These findings heralded the amyloid cascade hypothesis, which stipulates that the accumulation of Aβ is the main driving force behind the development of AD (6). Additional genetic evidence provides further support for the amyloid cascade hypothesis with the discovery that patients with APP gene duplications and Down syndrome develop Aβ plaque and AD-like pathologies (7, 8).
The mutations associated with early-onset AD quickly found their way into transgenic mouse models, and the findings from these models have largely supported the amyloid cascade hypothesis (9). Transgenic mice overexpressing various mutant APP constructs develop Aβ pathology with some also displaying cognitive decline reminiscent of human AD (10–12). Aβ deposits in progressive fashion and eventually develops into extracellular plaques. Similarly, transgenic APP mice exhibit impairments in behavioral tasks. In contrast, mutations in PS1 that cause FAD in humans do not lead to amyloid deposition and impaired behavioral phenotype in mice despite disruptions in calcium homeostasis and increases in Aβ1-42, a form of Aβ more prone to aggregation (13, 14). However, crossing APP and PS1 mice exacerbates the Aβ pathology and accelerates the development of cognitive impairments (15, 16). Thus, the introduction of multiple transgenes into mice has expanded our understanding of the role that mutations in PS1 and the subsequent increase in Aβ1-42 might play in the development of AD. Even though multiple mutations are not expressed in humans, the introduction of multiple transgenes in mice does provide relevant insight into the pathogenesis of AD.
Despite the introduction of the APP and PS1 transgenes in an attempt to better recapitulate AD pathology, these mice still lack tau pathology. Hence, it became obvious that more aggressive approaches would be necessary, including the introduction of additional transgenes to mimic both Aβ and tau pathology in a mouse. One means of achieving this goal is to cross independent transgenic lines (17). Alternatively, Aβ and tau pathology can be induced by injections of pathological protein (18). Although both of these approaches create models that mimic the Aβ and tau pathology found in AD, they pose certain practical difficulties such as poor breeding, the appearance of motor deficits unrelated to AD, and short life expectancy. These unfortunate constraints preclude the use of these models in various applications including learning and memory studies.
3 The 3xTg-AD Mouse Model
To further study the interaction between Aβ and tau and their impact on AD pathogenesis, we sought to develop a transgenic mouse model that exhibits both Aβ and tau pathology. To achieve this goal, APP and tau transgenes were co-microinjected into single-cell embryos from mice with a knock-in of PS1 with a M146V mutation (PS1M146V-KI) (19). The Swedish mutation of APP (APPswe) containing the double mutation KM670/671NL was used to induce the development of Aβ pathology. To ensure the development of pathological tau lesions, instead of using wild-type tau, a mutant form of tau associated with frontotemporal dementia with parkinsonism-17 (FTDP-17) was used. In FTDP-17 patients, tau deposits into NFTs similar to AD, but the deposits develop in different brain regions (frontal-temporally). Likely as a result of the spatial positioning of pathology, unlike AD where memory deficits are first observed, in FTD, executive functioning is impaired and behavioral symptoms (personality changes) are observed (20). Nevertheless, mutations in tau generally cause tau to aggregate more easily. Hence, the human tauP301L transgene was included in the 3xTg-AD mice.
Both the APPswe and tauP301L transgenes were under the control of the Thy1.2 promoter, which limits expression of transgenes largely to the CNS (21). These transgenes were co-microinjected into single-cell embryos from PS1M146V-KI mice. As noted before, the inclusion of the PS1 mutation in mice increases the production of Aβ1-42 and accelerates the development of pathology. Thus, with the 3xTg-AD mouse model, Aβ and tau pathology accumulates rapidly, allowing study of both pathologies in the mouse lifespan. Yet, pathology still appears progressively and in relevant brain regions, making the study of different stages of the disease possible.
In addition to the development of relevant AD-like pathology, the 3xTg-AD mice boast excellent colony-maintenance characteristics. Both the APPswe and tauP301L genes integrated into a single locus and sort as if a single gene. In addition, PS1M146V is “knocked-in” into the endogenous locus so essentially the mice breed as if they contained only a single transgene, despite harboring three transgenes.
3.1 Aβ Pathology in the 3xTg-AD Mice
As noted before, Aβ and tau pathology develop progressively and in a brain-region-specific manner in the 3xTg-AD mouse model. Thus, tracking the development of pathology added support to the amyloid cascade hypothesis. Despite equal transgene expression of both APP and human tau, accumulation of Aβ precedes tau accumulation in the 3xTg-AD mice, and it is likely that Aβ accumulation drives the development of tau pathology (19).
Aβ pathology in the 3xTg-AD mice first appears intraneuronally at 3–4 months of age in the neocortex. At this point in time, these accumulations are detectable by various anti-Aβ antibodies including Aβ1-42-specific antibodies, and most of the Aβ is still detergent-soluble. That soluble intraneuronal Aβ accumulation precedes plaque formation is consistent with other transgenic mouse models (22). Similarly, brain affected by Down syndrome shows evidence of increased Aβ production likely due to the increase in APP copy number and also forms intraneuronal Aβ deposits before extracellular accumulations develop (23). As the mice age, Aβ continues to accumulate as levels of both Aβ1-40 and Aβ1-42 increase. By 6 months, intraneuronal Aβ can be found in the hippocampus in the CA1 region and also in the amygdala. By 6 months, some of the intraneuronal Aβ is oligomeric in nature as the deposits are detectable by the antibody A11 (24).
Around 9 months of age, extracellular deposits of Aβ first begin to appear in the cortex. As extracellular plaques of Aβ develop and enlarge in size and number, intraneuronal Aβ begins to recede, but exactly what triggers the shift of Aβ from intracellular stores to extracellular space is not clear. Nevertheless, Aβ continues to accumulate extracellularly such that by 12 months of age, extracellular deposits can be seen in the hippocampus and some plaques become thioflavin-S positive, indicating the development of β-pleated sheet characteristics (25). By 15 months of age, extracellular plaques are widespread throughout the hippocampus and cortex. At this stage, large amounts of Aβ are found in detergent-insoluble fractions and likely fibrillar forms of Aβ dominate. However, even with the development of plaques, oligomeric forms of Aβ and other soluble Aβ species persist (24).
3.2 Tau Pathology in the 3xTg-AD Mice
Similar to the deposition of Aβ, tau pathology also follows a temporal and brain region-specific manner. Immunoreactivity for the antibody HT7, which detects human tau, is evident only in the somatic-dendritic compartment in pyramidal neurons of the CA1 region as early as 4 months of age (26). Mimicking the pattern observed in human brains, tau later spreads to cortical regions (19). As the mice age, reactivity with the conformation-specific MC-1 antibody appears first, followed by phosphorylation of tau. The MC-1 antibody detects a pathological conformation state of tau protein observed in the early stages of AD brains (27). The phosphorylation of tau at certain epitopes can be detected using monoclonal antibodies against specific phosphorylation sites. For example, the AT180 antibody detects tau phosphorylated at Thr231, AT8 detects tau phosphorylated at Ser202 and Thr205, and PHF-1 detects tau phosphorylated at Ser396 and Ser404. In the 3xTg-AD mice, phosphorylation at these sites appears in a sequential manner. Site-specific phosphorylation of tau at the AT180 and AT8 occurs before immunoreactivity with PHF-1 is observed (19). Although AT180 and AT8 immunoreactivity is apparent at 10–15 months of age, PHF-1 is only apparent at 15+ months of age. Tau accumulation is also apparent by Gallyas silver staining and Thioflavin-S at 12 months of age indicating that tau is depositing into aggregates (19). The presence of NFTs was also confirmed by electron microscopy analysis (Fig. 1).
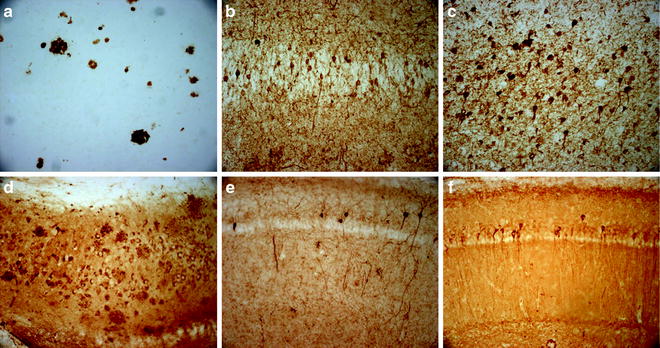
Fig. 1.
Representative AD pathology in the 3xTg-AD mice. (a) 6E10 immunoreactivity in 6-month-old 3xTg-AD mice reveals intraneuronal Aβ pathology in the subiculum; (b) 6E10 immunoreactivity in 15-month-old 3xTg-AD mouse shows the development of extracellular plaques; (c) 1560 immunoreactivity in human AD brain of extracellular Aβ plaques; (d) HT7 immunoreactivity in 6-month-old 3xTg-AD mouse brain reveals intraneuronal tau accumulation in CA1 region of the hippocampus; (e) HT7 immunoreactivity in 12-month-old 3xTg-AD shows increased tau accumulation with age; (f) AT8 immunoreactivity in human AD brain showing tau phosphorylation; (g) AT8 immunoreactivity first appears in 3xTg-AD mice at 10–12 months of age in the hippocampus; (h) PHF-1 immunoreactivity follows at 15+ months of age; (j) PHF-1 immunoreactivity is apparent in human AD.
3.3 Synaptic and Cognitive Deficits First Appear Alongside Intraneuronal Aβ Deposition
Synaptic deficits are the best correlate of the memory deficits in AD (28). Although plaque number does not correlate well with cognitive impairments, various Aβ species induce synaptic dysfunction. For example, Aβ oligomers induce LTP deficits (29). Synaptic dysfunction and impaired LTP is observed as early as 6 months in 3xTg-AD mice at a time when oligomeric Aβ is readily apparent (19). Furthermore, at this time point, Aβ oligomers appear intracellularly, indicating a possibility that these intraneuronal accumulations of Aβ contribute to the synaptic dysfunction.
As expected from the early synaptic dysfunction observed, behavioral deficits are apparent in 3xTg-AD mice at an early age. By the time synaptic dysfunction is apparent at 6 months of age, behavioral deficits manifest as retention impairments in the Morris water maze task (30). Fear conditioning is also impaired at this age. These impairments correlate with the appearance of intraneuronal Aβ in the hippocampus and amygdala, brain areas involved in memory and fear conditioning respectively. Thus, it is likely that intraneuronal Aβ plays a role in early behavioral impairments. This finding, however, does not preclude extracellular Aβ also contributing to the development of cognitive dysfunction. Notably, even as mice age and intraneuronal Aβ deposition wanes and gives way to extracellular deposits, behavioral deficits increase, and expand to include impairment in learning and memory (31).
3.4 Inflammation in the 3xTg-AD Mice
Inflammation is an invariant feature of the AD brain and is believed to be in response to the accumulation of fibrillar Aβ. Reactive microglia and astrocytes localize around Aβ plaques in the AD brain (32, 33). These activated glia are believed to be phagocytosing Aβ in an attempt to reduce pathology (34, 35). Activated microglia first appear around extracellular Aβ deposits around 12 months of age (26). The spatial and time-related appearance of activated microglia with fibrillar deposits of Aβ indicates that microglia activation may be induced by fibrillar Aβ. However, there are indications that inflammatory processes begin earlier. Elevated levels of tumor necrosis factor-α (TNFα) and monocyte chemoattractant protein-1 (MCP-1) were found in the entorhinal cortex at 6 months of age (36). Furthermore, primary microglia can be activated by monomeric and oligomeric forms of Aβ, which are present well before the appearance of extracellular Aβ deposits at 12 months (37, 38). Thus, it is likely that inflammatory processes in the AD brain occur throughout the disease process and may even contribute to the progression of the disease.
Even while activated glial cells are clearing Aβ, they are releasing various inflammatory mediators such as cytokines and chemokines that may actually be promoting pathology. Indeed, numerous cytokines have been shown to be elevated in the AD brain (39–42). These cytokines may play various roles in driving AD pathology including inducing tau phosphorylation. Cytokines have been shown to be able to induce tau phosphorylation in mice (43). Similarly, lipopolysaccharide (LPS)-induced increases in inflammation in the 3xTg-AD mice did not alter Aβ pathology, but tau phosphorylation was increased via a Cdk5-dependent mechanism (26). In addition to increases in tau phosphorylation, increased microglial activation was observed along with increases in Interleukin 1β (IL-1β). It is likely that the increased cytokines in the LPS-induced inflammatory state are responsible for the increase in tau phosphorylation. In the AD brain, Aβ accumulation induces inflammation and the subsequent increases in cytokines observed in AD brains may contribute to the development of tau phosphorylation. Thus, inflammation represents a pathway by which Aβ might induce tau pathology.
3.5 Modulation of AD Pathology in the 3xTg-AD Mice
In addition to modeling various aspects of AD, mouse models represent an important platform for the ultimate goal of developing or identifying treatments for AD. Preclinical trials of various treatments in the 3xTg-AD mouse model allow evaluation of the efficacy of reducing both Aβ and tau pathology. In this section, we will discuss several therapeutic treatments that modulate Aβ and/or tau pathology in the 3xTg-AD mice, and their limitations and potentials for treating AD in humans.
As described above, the genetic and pathological evidence suggests a central role for Aβ in the pathogenesis of AD (44). The work supporting the amyloid cascade hypothesis is compelling enough such that attempts to either reduce production of or increase degradation of Aβ have become prominent goals in the treatment of AD. One proposed method of reducing Aβ load is the direct removal of Aβ by vaccination or immunotherapy. The first Aβ immunotherapy was described in 1999, and the treatment was effective in reducing Aβ plaques in APP transgenic mice (45). Subsequent Aβ immunotherapy in other mouse models has shown similar effectiveness in reducing Aβ load and restoring behavioral impairments (46, 47). To study the efficacy of Aβ immunotherapy as a treatment for clearing both Aβ and tau pathology, antibodies against Aβ were administered to 3xTg-AD mice. Immunotherapy against Aβ was effective in clearing Aβ plaques, early intraneuronal Aβ deposits, and early tau aggregates from the brains of 3xTg-AD mice, as well as rescuing cognitive deficits (30, 48). Interestingly, Aβ immunotherapy does not clear hyperphosphorylated tau aggregates indicating that later tau aggregations are more permanent (48). In addition to providing evidence that Aβ immunotherapy may be useful for treating AD, immunotherapy has provided further evidence for the amyloid cascade hypothesis because tau is cleared even though immunotherapy is directed only against Aβ. Furthermore, after clearance of Aβ and tau using Aβ immunotherapy, pathology reemerges in a hierarchical fashion with Aβ deposition occurring first, followed by the reemergence of tau accumulation (48). Thus, Aβ accumulation appears to be able to induce tau accumulation in the 3xTg-AD mice.
One pathway by which Aβ may influence tau accumulation may be by inducing proteasome dysfunction. Proteasome activity can be a major clearance mechanism for tau protein (49). As noted above, Aβ immunotherapy is able to also clear tau pathology. Further studies showed that clearance of tau pathology with immunotherapy is mediated by the proteasome (48). In addition, hyperphosphorylated tau is not cleared by Aβ immunotherapy, and this is consistent with studies showing clearance of unphosphorylated tau by the proteasome system (50). Moreover, proteasome function has been shown to be directly impaired by Aβ oligomers in the 3xTg-AD mice (51). These results show that Aβ likely drives tau pathology by impairing proteasome function, leading to tau accumulation. Clearance of Aβ using immunotherapy can reverse this proteasome dysfunction leading to a restoration of tau aggregation. Clinical trials using Aβ immunotherapy have shown promise with indications of removal of Aβ in patients (52). However, clinical trials were stopped because 6% of patients developed subacute meningoencephalitis (53). Nevertheless, work continues on developing safer immunotherapy approaches against Aβ that will not induce dangerous side effects (54). For example, a novel gene therapy using a herpes simplex virus delivering Aβ1-42 and Interleukin 4 (IL-4) induces a T-helper 2 (Th2) antibody response that leads to clearance of Aβ pathology and decreases in tau phosphorylation in the 3xTg-AD mice (55). With this new form of immunotherapy, improvements in the Barnes maze task were observed, and new techniques such as this novel gene therapy may help clear pathology and treat AD.
In addition to immunotherapy, several pharmacological approaches have been used as treatments for AD. In particular, the acetylcholine receptor has been an important target for the treatment of AD. Acetylcholine receptors are divided into two major subgroups: nicotinic acetylcholine receptors (nAChR) and muscarinic acetylcholine receptors (mAChR). Selective loss of the nAChR along with a loss of cholinergic neurons is a notable and invariant feature in the AD brain (56, 57). Treatments for AD have focused on restoring acetylcholine levels in the AD brain and, until recently, the only major treatment for AD was the administration of acetylcholinesterase inhibitors. Because nAChR are selectively lost in the AD brain, the chronic use of nicotine in cigarette smokers has been studied as a risk factor for AD. Although epidemiological studies surrounding the use of nicotine and the risk of AD have provided nebulous results, some evidence exists that suggests that nicotine may be able to modulate Aβ and tau pathology differentially (58, 59).
Studies in transgenic mouse models of AD have given more insight into the role nicotine might play in AD pathogenesis. In APP transgenic mice, chronic nicotine administration led to decreases in Aβ plaque load, although soluble Aβ levels were unchanged (60). Unfortunately, effects of nicotine on tau pathology could not be studied because of lack of tau pathology in these mice. In contrast, the 3xTg-AD mouse model provides an opportunity to study nicotine and its effects on tau pathology. In addition, the 3xTg-AD model mimics the loss of α7nAChR that is observed in human AD and provides a good model for observing nicotinic effects on AD pathology (61). Similar to the studies in APP mice, administration of nicotine to 3xTg-AD mice did not alter soluble Aβ pathology (61). In contrast, increases in tau phosphorylation via activation of p38-mitogen-activated- protein kinase pathway was observed (61). Thus, nicotine may play a role in driving tau pathology in AD.
In contrast to the nicotinic receptors, mAChR are preserved in the AD brain, and they present an enticing target for restoring cholinergic hypofunction in AD (62). The M1 mAChR subtype, the predominant mAChR in the CNS, is involved in short-term memory processes, and represents a promising target (63). In addition, M1 agonists have shown ability to modulate both Aβ and tau pathology (64). However, their clinical use has been limited because of poor selectivity for the M1 receptor, poor bioavailability, poor activity, and narrow safety margin (63). The development of newer, highly selective M1 agonists presents an opportunity to study the effects of M1 agonists on AD pathology (65). The new M1 agonist AF267b was administered to the 3xTg-AD mice and was effective in reducing Aβ pathology and reducing tau phosphorylation in the hippocampus and cortex, but not in the amygdala (66). Accordingly, AF267b restored cognitive impairments on cortical and hippocampal-dependent tasks but impairments in contextual fear conditioning persisted (66). The M1 agonist was able to exert these effects by activating disintegrin and metalloproteinase 17 (ADAM17), shifting APP processing toward a nonamyloidogenic pathway, thereby reducing Aβ. The reduction in tau phosphorylation was mediated by a reduction in glycogen synthase kinase-3b (GSK-3b) activity. Because reducing GSK-3b activity was effective in reducing tau phosphorylation, lithium, a known GSK-3b inhibitor, was administered to the 3xTg-AD mice. Lithium is commonly used to treat bipolar disease, but because of its inhibitory activity against GSK-3b, it may have other uses. In 3xTg-AD mice, lithium was able to reduce tau phosphorylation, but Aβ load was not altered and cognitive function was not restored (67). Thus, it appears that reducing tau pathology with lithium is likely not sufficient to restore cognitive deficits.
Because direct application of Aβ induces synaptic dysfunction, clearance of Aβ is likely needed to restore cognitive function (68, 69). However, as noted before, reducing Aβ pathology often leads to reduced tau pathology. Immunotherapy leads to clearance of early tau pathology, whereas M1 agonists reduce tau accumulation (48, 66). Thus, although various direct applications of Aβ in vitro induce synaptic dysfunction, reductions in tau may also be responsible for rescuing cognitive deficits. Indeed, reductions of soluble tau and Aβ are necessary to restore behavioral function in 3xTg-AD mice (70). In contrast, reducing soluble Aβ alone is insufficient to rescue cognitive function. Furthermore, the presence of tau may facilitate Aβ-induced cognitive dysfunction. Reductions in endogenous tau levels can prevent cognitive impairments typically seen in transgenic APP mice (71). Thus, reduction of tau pathology may be in an important therapeutic target for treating AD.
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


