Chapter 23 Shock Syndromes
Definition of shock
A true understanding of shock syndromes must begin with the definition of shock. Shock is not defined by tachycardia, hypotension, circulatory collapse, stupor, coma, pale mucous membranes, or dehydration. These clinical signs may be associated with shock and are easily recognized, but they are common to many other conditions. The underlying problem or inciting event for all causes of shock is a relative insufficiency of intracellular energy production. Most often this is caused by a decrease in effective blood flow and oxygen delivery to tissues that results in failure to meet the demands of the tissues. Stated differently, this form of shock is “the state in which profound and widespread reduction of effective tissue perfusion leads first to reversible and then, if prolonged, to irreversible cellular injury.77” The decrease in effective perfusion can occur by many mechanisms, either cardiac or vascular in nature.
Classification of shock syndromes
In this chapter classification of shock syndromes will be divided into circulatory causes, hypoxemic causes, and metabolic causes. Circulatory causes can be further divided into hypovolemic causes, cardiogenic causes, and distributive causes. Table 23-1 lists examples of each classification.

Although such categorical organization of shock syndromes is helpful to understand the complex nature of shock, it is crucial to recognize that most clinical forms of shock may encompass several classifications. The utility of anatomic or functional categories of shock is questionable when viewed from a clinical perspective, and some have argued that such a classification is misleading because clinicians may approach a one-dimensional, easy-to-understand representation of shock with a simplistic one-dimensional approach to therapy.130 Unfortunately, shock is complex; it does not begin with a common pathophysiologic event and does not necessarily end with survival after a simple universal initial treatment. Each cause of shock sets in motion a complex series of events that include neural and hormonal responses, as well as numerous inflammatory cascades.
Additional disagreement also centers on the use of the term distributive shock. The term distributive has been used to describe various types of high-flow shock under the assumption that blood flow is not normally distributed to tissue beds. Used in this context, “distributive shock” is a theoretical designation of a type of shock syndrome that is not defined by criteria that can be easily measured in a clinical setting. Maldistribution of or heterogeneous blood flow has been documented in people in clinically accessible microscopic vascular beds (e.g., mucous membranes, sclera, liver, nail bed). Direct observations have documented the phenomenon of maldistribution of blood flow, but such measurements are not necessarily representative of all areas and are not quantitative measures of the extent of maldistribution throughout the body. Therefore maldistribution of blood flow is a physiologic concept that may be relevant to all shock states.130
Pathophysiology
Adequate tissue perfusion is not dependent on one simple value, rather it requires the integration of the entire cardiovascular system and is best described by effective circulating volume. Effective circulating volume (ECV) is a difficult to define term that describes the “fullness” of blood vessels. In essence adequate ECV requires an adequate blood volume delivered at an adequate pressure.11 Although tissues require the maintenance of ECV for long-term function, the cardiovascular system considers the maintenance of normal mean arterial pressure (MAP) as its number one priority.55 This is primarily because perfusion of the heart and the brain is pressure dependent; these vital organs need a minimum MAP for adequate perfusion. In terms of physics, maintaining a constant pressure is far more feasible for the cardiovascular system than maintaining a constant flow or volume.11Figure 23-1 outlines the interrelationship of the major cardiovascular parameters and how they influence MAP.
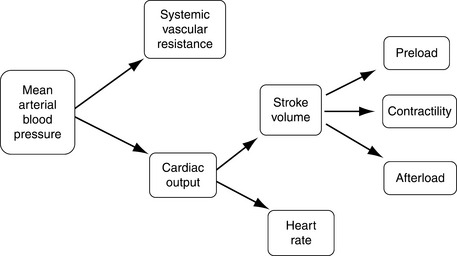
There are three main mechanisms by which decreases in ECV can occur (Table 23-2).61
1. Hypovolemia may lead to inadequate blood volume and hence inadequate preload, causing a decrease in cardiac output.
2. Cardiogenic causes in which abnormalities in cardiac function lead to inadequate cardiac output despite a normal or increased blood volume.
3. Distributive causes in which blood volume and cardiac function are normal but there are alterations in systemic vascular resistance globally or locally leading to inadequate ECV.

Hemorrhagic shock
Baroreceptor-Mediated Responses
When ongoing hemorrhage occurs in the conscious dog there is an initial normotensive period (moderate hemorrhage) followed by hypotension (severe hemorrhage).53,124 During moderate hemorrhage, the decrease in cardiac output is sensed by the high pressure baroreceptors of the carotid bodies and aortic arch. The subsequent decrease in baroreceptor afferent traffic to the vasomotor center of the brain causes sympathetic tone to increase, parasympathetic tone to decrease, and vasopressin release. This results in an increase in heart rate and systemic vascular resistance. Sympathetic mediated vasoconstriction is more prominent in precapillary arterioles and the blood vessels of the skin, skeletal muscles, and splanchnic viscera are vasoconstricted to a greater degree than the rest of the body. This is an effort to centralize blood volume and maximize perfusion of vital organs during the acute insult. Tachycardia and increases in systemic vascular resistance will contribute to the maintenance of a normal MAP in the face of volume loss (see Figure 23-1).41,53,124 This phase is also known as compensatory shock. There does appear to be some species variability in the nature of the hemodynamic responses to circulatory shock. In the dog it has been reported that there is little change in left ventricular contractility in association with the increase in sympathetic tone.60 In addition plasma epinephrine is not elevated during this normotensive hemorrhage in dogs.39
The renin-angiotensin system (RAS) is also stimulated by hemorrhage and the associated decrease in cardiac output. Renin release will be activated by the increase in sympathetic tone occurring as part of the baroreceptor response. In addition the afferent arteriole of the kidney is itself a baroreceptor and may sense the decrease in ECV directly. Plasma renin cleaves the plasma protein angiotensinogen, generating angiotensin I. Angiotensin I is rapidly converted to angiotensin II by angiotensin converting enzyme, which is present in the endothelium of the lung. Angiotensin II is one of the most potent vasoconstrictors of the body contributing to increases in systemic vascular resistance. Angiotensin II also stimulates the release of aldosterone. Both angiotensin II and aldosterone stimulate renal sodium retention in an effort to augment blood volume (see Chapter 3).11,55 As mentioned above, carotid sinus baroreceptor off-loading also stimulates the release of vasopressin (antidiuretic hormone) from the hypothalamus, which causes vasoconstriction and renal water conservation. Baroreceptor responses mediate changes on a minute-to-minute basis and are vital to surviving an acute injury or insult. The RAS responses take 10 minutes to an hour to have benefit and are more important in attempting to return the system back to the preinjury state and recovery.53
The pathophysiology above explains the six classic clinical signs of hypovolemic shock.
1. Decreased mentation due to inadequate perfusion of the brain.
2. Pale mucous membranes as a result of arteriolar constriction and decreased blood volume in the capillary beds.
3. Prolonged capillary refill time because the vasoconstricted arterioles delay the return of blood to the capillaries of the mucous membranes.
4. Tachycardia as part of the sympathetic, compensatory response.
5. Poor pulse quality due to vasoconstriction and decreased stroke volume.
6. Decreased extremity temperature compared with the core body temperature as a result of peripheral vasoconstriction.
Moderate, normotensive hemorrhage in experimental dogs occurs with the loss of less than 30% of blood volume. As the degree of hemorrhage increases there is a precipitous decrease in MAP and heart rate. In experimental dog studies, this usually occurs when following the removal or loss 30% of blood volume. This hypotensive phase is associated with central inhibition of sympathetic outflow and the animals cannot be salvaged despite administration of the shed blood and additional volume support.41,124 This is also known as decompensated or irreversible shock. Although decompensated shock is marked by bradycardia, the hypotension is independent of the bradycardia because it does not improve with atropine-induced tachycardia. The activity of the RAS continues to increase during hypotensive hemorrhage. In some species, including cats, rabbits, and rats, there is evidence that the sympathoinhibition seen with severe hemorrhage is stimulated by cardiac or cardiopulmonary receptors and transmitted via the vagus nerve.41 In contrast there is no apparent role of cardiopulmonary receptors in the generation of sympathoinhibition in dogs, the cause of the failure of sympathetic-mediated vasoconstriction in uncompensated shock in dogs is currently unknown.129 Additionally, there may be a relative vasopressin deficiency following prolonged shock states, further exacerbating the hypotension.40,78 Cardiac output and arterial blood pressure fall to zero with loss of 35% to 45% of total blood volume and is rapidly fatal.53
The clinical hallmark of decompensated shock is bradycardia and for this reason bradycardia (not due to conduction disturbances) may be a poor prognostic indicator in canine patients presenting in circulatory shock. In contrast bradycardia (or inappropriate normocardia) is not uncommon in cats having circulatory shock and does not appear to carry any prognostic significance in this species. The mechanism of bradycardia in hemodynamically unstable cats is unknown. It can occur in mild to moderate cases of shock and the heart rate generally increases as the animal is resuscitated.13,122 Hypothermia is recognized to cause bradycardia and is common in feline patients with circulatory shock, raising the possibility that body temperature could play a role in this phenomenon.107
Chemoreceptors
The chemoreceptors are specialized cells that sense decreases in oxygen, increases in carbon dioxide, and increases in hydrogen ion concentration. The peripheral chemoreceptors are found in the carotid body and aortic arch adjacent to the baroreceptors. When ECV decreases to a critical level, these cells are stimulated by the lack of oxygen and the accumulation of carbon dioxide and hydrogen ions. They transmit signals to the central vasomotor center in a manner similar to the baroreceptors, causing increases in sympathetic tone and vasopressin levels. They also stimulate the respiratory center, leading to increases in alveolar ventilation that maybe evident as tachypnea in the patient with circulatory shock. These receptors contribute to the maintenance of blood pressure in the face of severe decreases in ECV and are not thought to contribute to the regulation of MAP in the face of mild to moderate insults.11
Starling Forces
Hemorrhagic shock will also lead to alterations in Starlings forces. Following acute hemorrhage there is a sudden drop in capillary hydrostatic pressure that promotes the movement of fluid from the interstitium to the intravascular space. Interstitial fluid can replace up to 75% of the shed blood volume.10 This process is known as transcapillary refill or autotransfusion. As the protein concentration (and red blood cell concentration) of interstitial fluid is lower than that of blood, this response causes a decrease in both hematocrit and total plasma protein. In dogs and cats splenic contraction can supplement hematocrit and the end result may be a proportionally greater drop in the total protein than that of the packed cell volume following hemorrhage.22
Cardiogenic Shock
Cardiogenic shock causes decreases in effective circulating volume despite a normal, or frequently increased blood volume (see Table 23-2). A decrease in cardiac contractility or diastolic filling will impair stroke volume. From Figure 23-1 it can be appreciated that the compensatory responses to this abnormality will be similar to those seen in hypovolemic shock, with increases in systemic vascular resistance and tachycardia. Hence the clinical signs of cardiogenic shock will be similar to those of hypovolemic shock, namely, pale mucous membranes, prolonged capillary refill time, poor pulse quality, and differences between extremity and core temperature. Causes of cardiogenic shock include myocardial failure as may occur with dilated cardiomyopathy or end stage valvular regurgitation. Cardiogenic shock secondary to decreased diastolic filling can occur with tachyarrhythmias, hypertrophic cardiomyopathy, or pericardial tamponade.147 Patients presenting in cardiogenic shock may or may not have concurrent congestive heart failure typified by pulmonary edema, pleural effusion, or ascites. For example end-stage valvular regurgitation cases are likely to have significant morbidity associated with their congestive heart failure and are now demonstrating evidence of poor perfusion. In contrast patients with malignant tachyarrhythmias may have no evidence of congestive heart failure at the time of presentation for cardiogenic shock.
Distributive Shock
When regulation of vasomotor tone is abnormal it can cause circulatory shock despite an adequate blood volume and normal cardiac function (see Table 23-2). Global decreases in arteriolar tone will cause decreases in ECV that is sensed by the baroreceptors, as described for hemorrhagic shock. But in this scenario there is a failure of compensatory vasoconstriction in response to increases in sympathetic tone and tachycardia is the sole compensatory response. Global vasodilatation is marked by hypotension that is unresponsive to fluid administration. On physical examination these patients can have red mucous membranes, rapid capillary refill times, tachycardia, bounding pulses, and warm extremity temperatures reflecting vasodilatation.61 Some causes of distributive shock such as sepsis can cause heterogenous changes in microcirculatory vasomotor tone leading to inadequate perfusion of some tissue beds while there is normal or possibly excessive perfusion of others. If microcirculatory abnormalities occur in the absence of generalized vasodilatation the patient may have normal global hemodynamic parameters while some tissue beds are suffering from hypoperfusion and inadequate cellular energy production (also known as cryptic shock).38 This is another form of distributive shock.
Pathologic consequences of shock
Cellular Hypoxia
When tissue oxygen supply is inadequate, either due to global decreases in blood flow or maldistribution of blood flow, oxidative metabolism is compromised and cellular function becomes dependent on anaerobic energy production. Glycolysis, the only source of cellular energy in an anaerobic environment, is extremely inefficient with only 2 mol of adenosine triphosphate (ATP) being produced from each mole of glucose. This represents approximately 3% of the potential energy in the glucose molecules. For a short period of time this limited anaerobic energy production may prevent cell injury and death. When 1 mol of glucose is metabolized by glycolysis it produces 2 mol of pyruvate, in anaerobic conditions most of the pyruvate is then converted to lactate.54 This conversion allows glycolysis to continue as it regenerates essential NAD+ and prevents the accumulation of pyruvate. Lactate production during anaerobic metabolism occurs in conjunction with an equimolar production of hydrogen ions as a consequence of concurrent hydrolysis of ATP (Figure 23-2).120 The result is the formation of lactic acid. Given its inefficiency, anaerobic metabolism is limited in its ability to maintain normal function. In situations of acute, absolute cellular hypoxia, such as asphyxiation, anaerobic metabolism can only support life for approximately 1 minute.
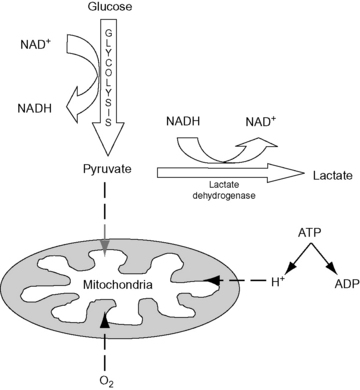
When inadequate cellular energy metabolism occurs, cell function is compromised. Maintenance of ionic gradients across the cell membrane requires active transport systems that consume 20% to 80% of all cellular energy produced.98 Some organs are more susceptible to hypoxic injury than others. The brain and the heart are obligate, aerobic, energy-dependent organs. Neurons use the majority of cellular energy in the preservation of ionic gradients and membrane potentials while myocardial cells have a high-energy requirement to fuel contractile processes. In the face of inadequate cellular energy production, the active transport systems controlling cell volume, such as the Na-K-ATPase pump, fail. As a consequence the entry of ions such as sodium and calcium into the cell is favored. In response to the increasing intracellular osmolarity, water shifts into cells leading to cell swelling and can ultimately result in cell death.17
Increases in intracellular calcium trigger activation of calcium-dependent phospholipases and proteases that can cause cellular injury. This includes calpainlike proteases that convert xanthine dehydrogenase to xanthine oxidase.134 Without sufficient levels of xanthine dehydrogenase, intracellular hypoxanthine accumulates. These changes have important repercussions during the reperfusion period.
Free Radical Damage
Reperfusion of organs following a period of ischemia, although essential for survival, can also be a mechanism of tissue damage. When oxygen is reintroduced to cells, it is used by xanthine oxidase (which accumulates during the ischemic period) to convert hypoxanthine (also accumulated during the ischemic period) to reactive oxygen species, such as the superoxide anion and hydrogen peroxide. These products will cause direct cell injury by damaging proteins and DNA and causing lipid peroxidation. Both rises in intracellular calcium concentration and plasma membrane damage can trigger activation of phospholipase A2, leading to arachidonic acid formation and eicosanoid synthesis including thromboxane A2 and leukotrienes.148 These arachidonic acid products have many functions including pro-inflammatory, procoagulant, and vasoactive effects.
Reactive oxygen species in turn have been shown to activate the nuclear transcription factor κB (NF-κB), which causes transcription of proinflammatory mediators, including more reactive oxygen species, leukocyte adhesion molecules, and tumor necrosis factor-α.94 These processes have also been shown to damage mitochondria such that cellular energy production may remain impaired, despite adequate oxygen delivery.16,47 This abnormality has been coined “cytopathic hypoxia” and is currently considered to be a contributor to the development of multiple organ dysfunction syndrome (MODS) in various disease states.83
Inflammatory Mediators
Cellular ischemia alters gene transcription including activation of NF-κB, which leads to cellular production of inflammatory mediators including tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β), IL-2, IL-6, interferon-γ, nitric oxide synthase, and cellular adhesion molecules.10,28,94 If this inflammatory response is substantial enough it will spill over into the systemic circulation (SIRS). When regulated, these responses are important to the maintenance of an effective immune response and tissue repair. An imbalanced or overzealous inflammatory response may result in global increases in capillary permeability, vasodilatation, leukocyte activation and adhesion, procoagulant changes, and mitochondrial dysfunction.47,79,82,88 Clinically this can result in the development of both hypovolemic and distributive shock. These mechanisms are potential contributors to acquired organ dysfunction following illness or injury.88
In addition to an exuberant proinflammatory responses, an antiinflammatory response (CARS) can also occur, which has been associated with immunocompromise and in some studies it correlates to a higher mortality.151 The development of CARS is associated with increased production of antiinflammatory cytokines, such as IL-10, IL-1 receptor antagonist, and transforming growth factor-β. Leukocyte inhibition and down regulation of NF-κB have also been demonstrated.1,2 There is some discussion in the current literature that CARS occurs concomitantly with SIRS as a normal response that limits the systemic inflammatory process.2 Although there is evidence that CARS maybe associated with an enhanced susceptibility to infection, this relationship is complicated and in many respects CARS may be a protective response. Following trauma, the immune system is activated and a second immune stimulus during this period may augment the initial response (second hit phenomenon) and potentially worsen the outcome.102,150 For this reason there may be a benefit in delaying major surgery, such as fracture repair, for at least 4 days following injury.
Inadequate perfusion of the gastrointestinal tract is common in circulatory shock because sympathetic responses tend to shunt blood flow toward vital organs and cause disproportionate splanchnic hypoperfusion.11 Gastrointestinal tract hypoperfusion has been shown to be an important contributor to posttrauma multiple organ dysfunction syndrome in experimental animals and people.99,143 Ischemia and reperfusion of the gastrointestinal tract can lead to production of inflammatory mediators and activation of neutrophils. This can amplify the inflammatory response occurring following cellular ischemia subsequent to shock and lead to severe SIRS and MODS. Bacterial translocation and subsequent bacteremia can also occur following a severe circulatory shock insult and further drives the development of severe SIRS.27 In summary the immune response to injury or illness is complex and yet to be fully understood. The cellular changes that occur following an insult have a protective role but in some circumstances they can become a source of harm to the patient. As clinicians the corner stones of therapy of systemic inflammatory responses are maximizing oxygen delivery to the cells and minimizing further systemic insults. Currently there are no recommended specific therapeutic agents to modulate the immune response.
Sequelae of circulatory shock
Animals that survive the acute episode of circulatory shock may still be at risk of developing SIRS, which, if severe, may result in MODS and possibly death in the days following the original insult or injury. The likelihood of SIRS and MODS following circulatory shock will increase with increasing severity and duration of the shock episode.28 In human medicine, other independent predictors of MODS following trauma include male gender, elderly patients, amount of red blood cell transfusions, and the persistence of an elevated blood lactate concentration 12 to 24 hours postinjury.123
There is evidence in human medicine that certain functional genetic polymorphisms can influence patient mortality.96,103 For example variants of the tumor necrosis factor gene are associated with the occurrence of sepsis and death following trauma.96 Further the cellular responses to injury show sexual dimorphism. For example, in animal experimental models and human clinical patients estrogen plays a protective role following hemorrhagic shock or sepsis.10 The role of gender and genetic predisposition in the response to injury and outcome of veterinary patients is currently unknown.
Clinical management
Restoration of adequate circulation requires identification and control of any internal or external hemorrhage. Fluid therapy is the cornerstone of treatment for shock. Although fluid therapy is frequently contraindicated in patients with cardiogenic shock or hypervolemia, most other types of shock will be at least partially responsive to intravascular volume augmentation. Aggressive, yet judicious use of fluids will serve to increase tissue perfusion, decrease tissue hypoxemia, reduce secondary cytokine injuries, and maximize a successful outcome. A prospective study looking at people who died in the hospital after admission for treatment of injuries found that inadequate fluid resuscitation was the most common mismanagement recorded.25 Adequate intravascular volume replacement is crucial to restore perfusion to the major organs, thus reducing morbidity and mortality associated with hypovolemia. Approximately 50% of hypotensive, septic humans will have normalization of cardiovascular hemodynamics with fluid therapy alone.119
Access to the venous circulation is vital for rapid volume resuscitation. The intravenous or intraosseous routes are preferred because absorption from the subcutaneous or peritoneal space is slow and unpredictable, especially in the face of systemic vasoconstriction. Peripheral veins are preferred for the initial resuscitation efforts, but a jugular catheter may prove beneficial once the patient is more stable. The cephalic, lateral saphenous (dogs), or medial saphenous (cats) veins are most commonly used for initial placement of one or two intravenous catheters. The catheter(s) should be an over-the-needle catheter that is as large and short as possible to maximize flow rates through the catheter because the rate of flow is proportional to the radius to the fourth power and inversely proportional to the length. If venous catheter placement is not possible, either a venous cutdown or intraosseous catheter placement should be performed. Intraosseous catheter placement is further discussed in Chapter 15.
Once an intravenous catheter is placed, the clinician must decide what type and how much fluid to administer for the treatment of shock. There are basically four types of fluids that are typically used for the management of shock: crystalloids (isotonic and hypertonic), synthetic colloids, natural blood products (red blood cells, plasma, albumin), and oxygen carrying solutions. The various types and doses are listed in Table 23-3. Although the specific type of shock may help dictate the best therapeutic approach, it is important that the clinician understand the constituents of and potential side effects of each fluid type.
Table 23-3 Fluid Choices for Circulatory Support
| Fluid Type | Dose | Comments |
|---|---|---|
| Isotonic crystalloids | Dog: up to 90 mL/kg Cat: up to 60 mL/kg | Used in animals for intravascular and interstitial volume deficits. May cause edema in animals with capillary leak or a low oncotic pressure. |
| Synthetic colloid solutions (hydroxyethyl starches) | Dog: 5-20 mL/kg Cat: 5-10 mL/kg | Used in animals for volume replacement and oncotic support. May cause coagulopathies. |
| Human serum albumin | 2 g/kg or calculate albumin deficit (g): 10 × (desired-patient albumin) × weight (kg) × 0.3 | Used for albumin and oncotic support and volume replacement. Monitor closely for reactions. One time dose recommended. |
| Canine serum albumin | 1-2 g/kg/day | Limited safety studies thus far. Use with caution. |
| Fresh frozen plasma | 10-15 mL/kg as needed | Used to treat clotting factor deficiencies and provide albumin-containing oncotic support. |
| Frozen or cryo-poor plasma | Used to provide albumin-containing oncotic support or treatment of rodenticide toxicity. | |
| Packed red blood cells | 10-15 mL/kg to raise PCV by 10%-15% | Used to treat anemia. |
| Fresh whole blood | 20-25 mL/kg | Used to treat anemia, thrombocytopenia, clotting factor deficiencies, and provide albumin-containing oncotic support. |
Modified with permission from Silverstein DC. Daily intravenous fluid therapy. In: Silverstein DC, Hopper K, editors. Small animal critical care. St Louis: Saunders Elsevier, 2009.
Isotonic crystalloids
Isotonic crystalloids, also known as replacement fluids, are electrolyte-containing fluids with a composition similar to that of the extracellular fluid. They have a similar osmolarity as plasma and the electrolytes are small in size (i.e., sodium has a molecular weight of 23 Da compared with glucose at 180 Da). Examples include 0.9% sodium chloride, lactated Ringer’s solution, Normosol-R, and Plasmalyte 148. Although decades of investigation have not defined the ideal fluid for the treatment of shock, the initial resuscitation fluid for the treatment of patients in shock is most commonly isotonic crystalloids. A dose up to approximately one blood volume is typically used: 90 mL/kg in the dog and 50 mL/kg in the cat. Isotonic crystalloids rapidly distribute into the extracellular fluid compartment following administration, and only approximately 25% of the delivered volume remains in the intravascular space by 30 minutes postinfusion.131 Although theoretically this increase in interstitial fluid volume might predispose to interstitial edema and deranged oxygen transfer to the cells, this has not been shown in a canine hemorrhagic shock model.9 However, it is important that the veterinarian avoid overzealous use of isotonic crystalloids to prevent volume overload and interstitial edema, pulmonary edema, or cerebral edema. Patients with a low colloid osmotic pressure, pulmonary contusions, cerebral trauma, fluid nonresponsive renal disease, or cardiac disease are at highest risk for complications. In addition, substantial hemodilution of red blood cells, plasma proteins, clotting factors, and platelets can occur. Therefore, anemia, hypoproteinemia, and hypocoagulability should be anticipated following large-volume crystalloid administration.
Since it is hard to predict how a given animal will respond to a rapid fluid bolus, it is recommended that initially only one third to one half of the shock dose be given as quickly as possible (often using a pressurized fluid infusion system), followed by additional boluses as indicated by clinical parameters and repeated physical examinations. Animals with recently lacerated or ruptured blood vessels are susceptible to rebleeding following aggressive fluid therapy and a rapid increase in vascular hydrostatic pressure (and “pops the clot”). Hypotensive fluid resuscitation (to a mean arterial pressure of 60 mm Hg) may help prevent rebleeding while helping to maintain perfusion to vital organs.66
Not all isotonic fluids are created equal, as seen in Table 23-4. Isotonic saline solution (0.9% NaCl) contains a higher concentration of sodium and chloride compared with normal plasma, and will cause proportional changes (increases) in a normal animal’s electrolytes. Therefore, large amounts of 0.9% NaCl will cause a mild increase in sodium, a marked increase in chloride, and a moderate decrease in bicarbonate and potassium. The kidneys will typically compensate, if possible, by excreting the excess electrolytes and conserving potassium. Animals with hypochloremia, mild hyponatremia, or a metabolic alkalosis will often benefit from the administration of 0.9% NaCl.
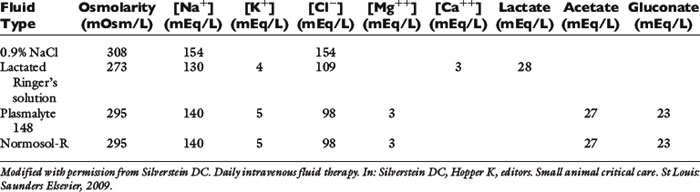
1. The isotonic crystalloid of choice for animals with head trauma is 0.9% NaCl, if possible, because this fluid has the highest sodium concentration and is therefore least likely to cause a decrease in osmolarity and subsequent water movement into the brain interstitium.
2. Animals with severe hyponatremia or hypernatremia should receive crystalloid fluids that most closely match the patient’s sodium concentration during resuscitation to prevent a rapid increase or decrease in serum osmolarity and subsequent central pontine myelinolysis (often delayed in onset) or cerebral edema, respectively.
3. Animals with severe liver disease should receive nonlactate fluids. Neonates may not be able to adequately metabolize the lactate and animals with diabetic ketoacidosis could have delayed clearance of ketones following lactate administration; however, there is a lack of evidence to support these theories.
4. Patients with a hypochloremic metabolic alkalosis may benefit from 0.9% NaCl because this is the highest chloride-containing fluid.
Synthetic colloids
Synthetic colloids are polydisperse solutions with large molecules (molecular weight >20,000 Da) that do not readily sieve across the vascular membrane. Most synthetic colloidal particles are suspended within a crystalloid base solution. These fluids are hyperoncotic to the normal animal and therefore cause the movement of fluid from the extravascular to the intravascular space. Intravascular oncotic pressure is primarily regulated by albumin (69,000 Da), and the normal colloid osmotic pressure (COP) in most small animal patients is approximately 20 mm Hg. Synthetic colloids lead to an increase in blood volume that is greater than that of the infused fluid volume and also aid in the retention of this fluid in the vascular space (assuming normal capillary permeability).131 Although there is no definitive evidence to support the use of colloids over crystalloids for the treatment of shock, they may have a longer intravascular effect, require smaller volumes to achieve similar intravascular volume expansion, and prove less likely to cause interstitial edema due to their hyperoncotic characteristics. However, their use is also associated with coagulation impairment, higher costs, and possible side effects (e.g., allergic reactions or renal impairment, both primarily reported in humans).
The primary synthetic colloid solutions available contain either dextrans, gelatins, hemoglobin-based oxygen carriers (HBOCs), or hydroxyethyl starches. Dextrans are composed of naturally occurring glucose polymers, but the most commonly used and studied dextran, dextran 70, is not currently commercially available. Gelatins are made following the hydrolysis of bovine collagen and subsequent succinylation or linkage to urea. The available gelatin, oxypolygelatin, has numerous side effects and a short duration of action, making it a less desirable synthetic colloid that is unlikely to gain widespread use. HBOCs contain stroma-free, ultrapurified hemoglobin glutamers that are highly polymerized to prolong their effect in the circulation. Hydroxyethyl starches (HES) are made from a wide size range of amylopectin polymers with variable chemical modifications that influence their pharmacokinetics and metabolism. These are the most commonly used synthetic colloids and will therefore be reviewed in detail. However, the characteristics of most available synthetic colloid solutions are displayed in Table 23-5.
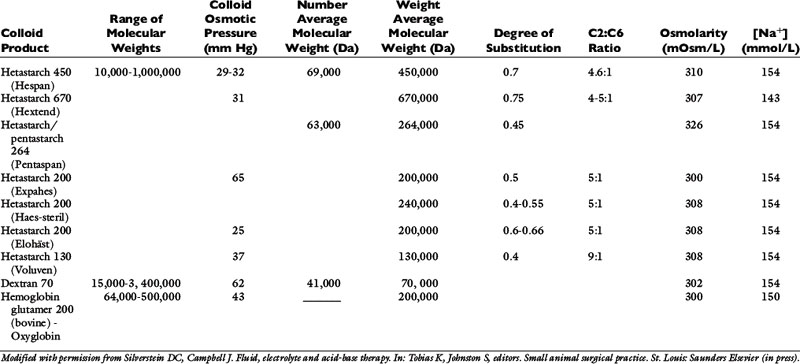
Examples of HES solutions include hetastarch, pentastarch, and tetrastarch (e.g., Voluven). HES preparations contain high polymeric glucose compounds that are manufactured by modification of the highly branched starch, amylopectin. Replacement of hydroxyl groups with hydroxyethyl groups at the C2, C3, or C6 carbon position of the constituent glucose molecules prevents rapid degradation by amylase. The ratio of substitution at the C2 versus C6 position (known as the C2:C6 ratio) also alters the half-life of the solution, with a higher ratio corresponding to a longer half-life. The degree of substitution (DS) refers to the number of hydroxyethyl groups per molecule of glucose and the higher the number of substitutions, the slower the breakdown and elimination of the molecule. However, a higher degree of substitution also means greater potential effects on coagulation.153 Hetastarch solutions have a rather high DS (0.6 to 0.7), while pentastarches and tetrastarches have a DS of 0.5 and 0.4, respectively. HES solutions are further characterized by their MW (low MW 70 kDa, medium MW 130 to 270 kDa, and high MW 450 kDa), their concentration (3%, 6%, or 10%), and their degree of substitution (0.4, 0.5, 0.6, or 0.7). It is important to note whether the MW is expressed as the number average molecular weight (MWn, most reflective of oncotic pressure) or the weight average molecular weight (MWw, exaggerated by larger particles). The MWw is determined by light scattering and is not as accurate a measure of the size of the colloid as MWn, which is the arithmetic mean of the range of molecular weights in the solution. The MWw is larger than the MWn, and as the molecular weight distribution of the colloid becomes narrower, MWw approaches and eventually equals MWn. In addition, the ability of synthetic colloids to modulate inflammation is related to their size and DS; those with a lower MW (<200 kDa) and DS (<0.4) may help to decrease capillary permeability, down-regulate the expression of adhesion molecules, inhibit neutrophil recruitment, and minimize cytokine production.46,84,145,146,159
Synthetic colloids are typically used in combination with isotonic crystalloids to maintain adequate plasma volume expansion with lower interstitial fluid volume expansion. Smaller total fluid volumes are needed and can therefore be administered more rapidly with fewer side effects. Measurements of COP are recommended with prolonged colloid administration. Animals with a colloid osmotic pressure less than 16 mm Hg may benefit from synthetic colloid administration and therapy should be adjusted to maintain values above this level. Animals with chronic hypoproteinemia may not need a COP greater than 16 mm Hg because the ratio of protein in the IV to interstitial space may not be as deranged as in those patients with acute IV losses. Total protein refractometer readings are not a valid means of monitoring colloid therapy.14
Potential side effects of synthetic colloid use are primarily related to disruption of normal coagulation. These include a decrease in factor VIII and von Willebrand factor concentrations (beyond just a dilutional effect), impairment of platelet function, and interference with the stability of fibrin clots, which makes them more susceptible to fibrinolysis.23,139,140,142,157 In addition, the time to clot formation (R), clot strength (maximum amplitude), and the speed of clot strength development (angle) using thromboelastography are all adversely affected by hydroxyethyl starches.45 The clinical manifestations of these changes are variable and depend on the status of the patient. Obviously, those patients with preexisting coagulopathies, von Willebrand disease (VWD), or moderate to severe thrombocytopenia/thrombocytopathia are at highest risk. Monitoring of the activated partial thromboplastin time (aPTT) may be helpful in assessing the adverse effects and risk level associated with the use of synthetic colloids, although there are no precise guidelines and it is difficult to predict which animals will develop clinical bleeding following administration. In general, the appropriate use of synthetic colloid solutions is deemed worth the risk, but judicious use of plasma and other blood products may also prove necessary to prevent bleeding complications, especially perioperatively. Caution should also be exercised to prevent volume overload or excessive hemodilution when large volumes of synthetic colloids are given to a patient. Additional side effects of synthetic colloids in people include renal impairment and allergic reactions, but similar problems in animals have not been documented.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


