Chapter 19 Fluid, Electrolyte, and Acid-Base Disturbances in Liver Disease
Normal physiology of the hepatobiliary system
Bile formation: composition and flow
Bile is an aqueous solution containing organic and inorganic compounds and electrolytes (Table 19-1).174 Separate hepatic and ductular transport mechanisms allow regulation of bile composition and volume in response to changing physiologic needs.110 Bile acids are amphipathic organic anions synthesized and conjugated by the liver. The hepatocyte is a polarized secretory epithelial cell with specific transporters localized in basolateral and canalicular cell membranes.142 The canaliculus is a confined space formed by a junction between specialized portions of cell membranes from two adjacent hepatocytes. The surfaces defining the canaliculus form a tight junction that functions as an anatomic barrier to solute diffusion. Transport processes in the basolateral hepatocellular and canalicular membranes determine bile acid uptake and biliary excretion. Active transport of osmotically active solutes into the canaliculus provides the driving force for bile flow.

Bile salts are the most concentrated organic solutes in bile and a major determinant of bile secretion. Rate-limiting secretory mechanisms involve bile acid transporters in the canalicular membranes. Bile acids impart unique properties that attenuate the osmotic forces in bile. Formation of bile acid micelles (polymolecular aggregates) protects the intestinal mucosa from highly concentrated solutes and promotes interaction between bile acids and lipids in the intestinal tract, thus facilitating digestion. Almost all bile acids are conjugated (exclusively to taurine in the cat and to taurine or glycine in the dog) and exist as organic anions rather than undissociated acids. Nonabsorbable constituents of bile (e.g., bile acids, phospholipids, cholesterol) are concentrated when water and inorganic electrolytes (e.g., sodium, chloride, bicarbonate) are absorbed from the gallbladder and biliary ducts. Stasis of bile flow or dehydration can promote a pathologic thickening of bile (inspissated or sticky consistency), whereas choleresis (increased bile flow) produces watery or dilute bile. The bicarbonate concentration of bile exceeds that of plasma and is largely under the influence of secretin. Most of the bicarbonate in bile arises during bile transport through biliary ductules. Bile formation and flow are driven mainly by osmotic mechanisms. Flow is initiated by bile acid-dependent and acid-independent mechanisms. In the basal state, equal contributions to flow are derived from canalicular bile salt-dependent and bile salt-independent mechanisms and from ductule processes. In the absence of bile salts, bile flow reaches only 40% to 50% of normal. Transcellular rather than paracellular mechanisms are most important in determining bile composition. Transcellular mechanisms concentrate bile acids and other solutes, whereas paracellular mechanisms permit simple diffusion (water and electrolytes) down electrochemical or osmotic gradients (Figure 19-1).
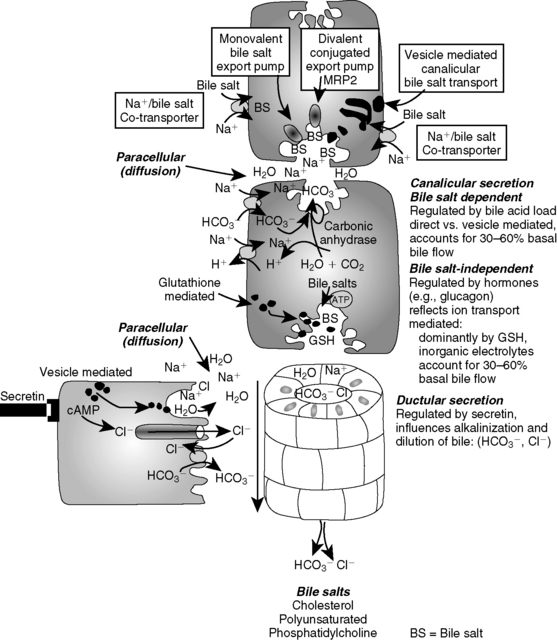
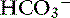 exchanger leading to bicarbonate secretion in ductal bile. Whereas bile formation occurs continuously, hormones (e.g., glucagon) can increase bile salt-independent mechanisms. Ductular secretions are stimulated by secretin causing bile alkalinization and dilution.
exchanger leading to bicarbonate secretion in ductal bile. Whereas bile formation occurs continuously, hormones (e.g., glucagon) can increase bile salt-independent mechanisms. Ductular secretions are stimulated by secretin causing bile alkalinization and dilution.There is a direct linear relationship between canalicular bile acid concentrations and bile flow. Non–micelle-forming bile acids (e.g., dehydrocholate) have the greatest effect. Hepatocellular uptake of bile acids is an energy-dependent process linked to sodium transport. This process accounts for approximately 80% of taurocholate uptake but only 50% of unconjugated cholate uptake.142 Protein carriers facilitate cytosolic transport of bile acids to canalicular membranes. Efflux of bile acids into canaliculi involves several mechanisms including facilitated diffusion dependent on canalicular carrier proteins, an adenosine triphosphate (ATP)-dependent mechanism, and exocytosis of cytosolic vesicles. Collectively, transcellular transport of bile acids and micelle formation maintain a marked concentration gradient between bile and blood, permitting biliary concentrations to exceed plasma bile acid concentrations by 100- to 1000-fold.
Bile ducts contribute to bile formation and modification as well as to bile flow. Production of ductular fluid primarily is under the influence of secretin, which regulates spontaneous or basal bile flow. Gastrin (but not pentagastrin) also increases bile duct secretion in dogs, whereas somatostatin decreases ductular bile flow. Increased ductular bile flow results in bile alkalinization and dilution. Disease states causing bile ductule proliferation also increase bile flow (e.g., cirrhosis, extrahepatic bile duct occlusion, inflammatory disorders). Bile ductules and ducts can also reabsorb bile as shown in cholecystectomized dogs.74
Hepatic nitrogen metabolism: detoxification, excretion, and role in acid-base balance
Urea Cycle and Glutamine Cycle
The liver converts waste nitrogen to an excretable form.55 Nitrogen derived from amino acids can be converted to ammonia directly or indirectly after incorporation into glutamate or aspartate in the liver. Ammonia subsequently is detoxified by conversion to urea (Figure 19-2). Two mechanisms exist for hepatic nitrogen detoxification. The hepatic urea cycle is best known and involves a linked series of enzymatic reactions carried out in the mitochondria and cytosol of the hepatocyte (see Figure 19-2). The second mechanism, the glutamine cycle, involves transport of glutamine into mitochondria, where it is converted to ammonia and used as a precursor of carbamoyl phosphate (see Figure 19-2). The urea cycle is a low affinity system, most important during alkalosis, whereas the glutamine cycle is a high affinity system, most important during acidosis. Collectively, these systems efficiently cleanse portal blood of ammonia. Approximately 25% of the ammonia for urea synthesis is derived directly from portal blood, and the remainder is derived from catabolism of proteins, peptides, and amino acids.
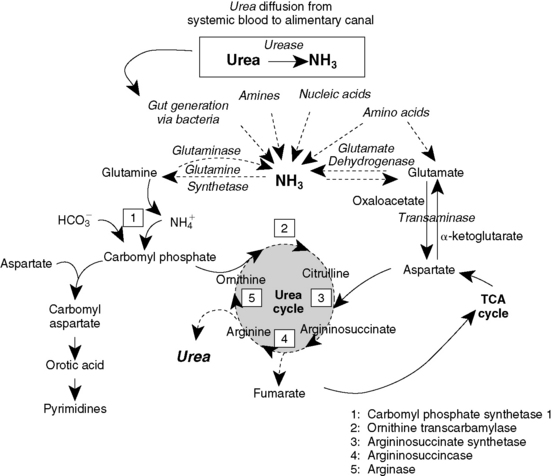
Urea synthesis depends on substrate supply, hormonal regulation, nutritional status, and liver cell volume. Regulation of urea cycle enzymes corresponds to the level of dietary nitrogen intake and possibly liver cell volume. The urea cycle may play an important role in acid-base homeostasis, as explained by the following reaction (using the amino acid alanine as an example of a nitrogen source)55:
Generation of one positive (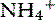 ) and one negative (
) and one negative (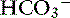 ) charge has the potential to maintain electroneutrality. However, because physiologic pH is in the range of 7.0 to 7.4, only 1% of ammonia exists as ammonia. Therefore the protons represented by the ammonium ions cannot be readily transferred to
) charge has the potential to maintain electroneutrality. However, because physiologic pH is in the range of 7.0 to 7.4, only 1% of ammonia exists as ammonia. Therefore the protons represented by the ammonium ions cannot be readily transferred to 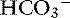 , and thus catabolism of large amounts of amino acids or protein can generate high bicarbonate concentrations resulting in metabolic alkalosis. Normally, detoxification of ammonia to electroneutral urea prevents changes in systemic pH55:
, and thus catabolism of large amounts of amino acids or protein can generate high bicarbonate concentrations resulting in metabolic alkalosis. Normally, detoxification of ammonia to electroneutral urea prevents changes in systemic pH55:
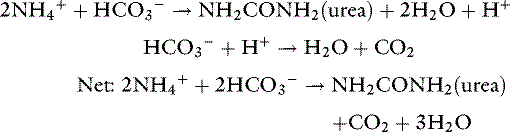
The preceding model probably is an oversimplification. Consumption of a diet composed of a complex mixture of amino acids (anionic, cationic, and sulfate-containing amino acids) results in a net gain of protons that must be excreted or neutralized. Urinary excretion occurs via dihydrogen phosphate (titratable acidity) and renal tubular production of ammonium from glutamine. Traditional concepts of renal tubular acid titration consider ammonium ion formation an important mechanism of acid-base regulation. However, ammonium ions excreted in urine are incapable of titrating acid because they are already protonated.55 An alternative view is that urinary excretion of 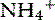 represents a mechanism by which the liver is deprived of substrates for urea synthesis, resulting in less bicarbonate neutralization and mitigation of acidosis. According to this hypothesis, the kidneys determine the route of nitrogen disposal, whereas the liver plays a more active role in systemic acid-base balance.
represents a mechanism by which the liver is deprived of substrates for urea synthesis, resulting in less bicarbonate neutralization and mitigation of acidosis. According to this hypothesis, the kidneys determine the route of nitrogen disposal, whereas the liver plays a more active role in systemic acid-base balance.
Serum proteins: albumin and globulins
Albumin
Albumin accounts for 25% of the proteins synthesized by the liver.151 Serum albumin concentration reflects the net result of synthesis by hepatocytes, systemic distribution, and degradation. Being relatively small in size (66,000 Da), albumin can be lost from the circulation through pathologically altered vessels (e.g., vasculitis), gut wall (e.g., lymphangiectasia), or glomeruli (e.g., glomerulonephritis, amyloidosis) or into the peritoneal cavity as a result of hepatic sinusoidal hypertension. Impaired or down-regulated hepatic albumin synthesis or losses exceeding synthetic capability result in hypoalbuminemia of variable severity. The liver has a tremendous reserve capacity for albumin synthesis.179 Normally, only 20% to 30% of the hepatocytes produce albumin, and synthesis can be increased as needed by a factor of 200% to 300%.75
Hepatic albumin production fluctuates depending on physiologic conditions and requirements (Figure 19-3). The most important variables are nutrition and interstitial osmotic pressure as sensed by the hepatocyte.178 The influence of nutrition on albumin production can be dramatic. Albumin synthesis decreases by 50% within 24 hours after a fast or with consumption of a protein-deficient diet. Serum albumin concentration reflects this change only after a lag period ranging from days to weeks as a new balance is achieved between exchangeable albumin pools. Feeding excessive calories in a protein-restricted ration augments development of hypoalbuminemia, as does dietary depletion of branched-chain amino acids.115,132,179 Hypoalbuminemia, caused in part by reduced albumin synthesis, also can be a consequence of changes in serum oncotic pressure related to hyperglobulinemia and treatment with synthetic colloids (e.g., dextran).66,178 Synthesis of albumin also decreases, sometimes dramatically, during critical illness as part of a negative acute-phase response.32,38
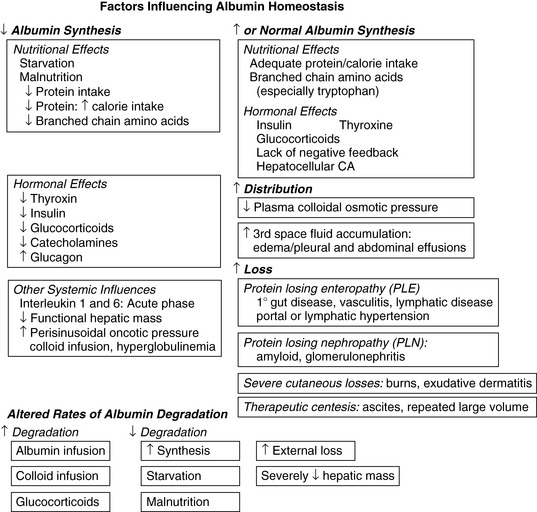
Hepatocellular synthesis of albumin is affected by a number of factors, the most important of which is the COP of the hepatic interstitial matrix.179 A decrease in COP stimulates albumin production whereas an increase in COP results in decreased albumin synthesis. After synthesis in the hepatocyte, albumin is released into the space of Disse by exocytosis. It then diffuses into the hepatic sinusoids, where it mingles with the systemic circulation. It then is dispersed into the interstitial space, returning to the systemic circulation via lymphatics and the thoracic duct. In normal animals, 50% to 70% of albumin is located extravascularly, with the largest amounts in interstitial spaces in skin and muscle.132 Normal transcapillary escape approximates 5% per hour, but inflammation may increase this several fold. This phenomenon commonly contributes to the “negative-acute-phase” effect that modestly lowers serum albumin concentrations in inflammation.
Catabolism of albumin probably occurs within or adjacent to vascular endothelium of tissues.241 The half-life of plasma albumin is 7 to 10 days in dogs and 6 to 9 days in cats.68,69,79 The rate of albumin catabolism is highly variable, but its fractional catabolic rate is directly proportional to the plasma albumin concentration and pool size.104 In conditions that cause hypoalbuminemia, the fractional and absolute rate of albumin catabolism decreases. The rate of albumin catabolism increases after albumin or synthetic colloid transfusion. Thus transfusion of albumin or infusions of synthetic colloids may potentiate endogenous hypoalbuminemia by two separate mechanisms. As a consequence of the large space of distribution and numerous mechanisms influencing the synthesis, distribution, and catabolism of albumin, serum albumin concentration does not accurately reflect contemporary changes in total body albumin resources or its hepatic synthesis.
The strong net negative charge of albumin (−17) explains its important contribution to the strong ion difference (SID) and allows it to bind weakly and reversibly with a variety of ions. In this capacity, albumin functions as a circulating depot and transport molecule for many ions (e.g., Ca2+, Mg2+, Cu2+) and metabolites (e.g., fatty acids, thyroxine, bilirubin, bile salts, amino acids).136 Albumin accounts for most of the plasma thiol content (i.e., sulfhydryl bonds) and provides protection against oxidative stress.175 Albumin also provides antioxidant activity by binding reactive transition metals (e.g., Cu2+) that catalyze free radical generation.136 Other important effects of albumin involve anticoagulant, antithrombotic, and antiinflammatory effects.
Oxidized and glycosylated forms of albumin occur in human patients with cirrhosis,231 and these forms increase in concentration as total serum albumin concentration decreases. The increase in the oxidized form of albumin reflects its role as a scavenger of reactive oxygen species. Glycosylation of albumin influences its binding and permeability characteristics and augments platelet aggregation, which may predispose to thromboembolic complications.231 The clinical implication of a lower reduced/oxidized albumin ratio lies in its relationship to oxidative stress imposed by low thiol substrate availability.
Numerous factors influence serum albumin concentration (see Figure 19-3). Modest hypoalbuminemia may reflect reduced albumin synthesis or enhanced catabolism, but these usually are slow in onset. Protein catabolism caused by illness usually spares albumin and targets muscle. The acute-phase response to tissue injury enhances transcapillary escape of albumin and may reduce lymphatic clearance. The most dramatic rapid reduction in serum albumin concentration is dilutional in nature and associated with crystalloid administration (with or without synthetic colloid). Such therapeutic dilutional effects typically aggravate acute severe extracorporeal losses (e.g., hemorrhage). Albumin loss resulting from protein-losing enteropathy or nephropathy initially is compensated for by albumin flux between intravascular and interstitial pools. With chronicity, a net body albumin deficit becomes apparent, and hypoalbuminemia develops. The most severe chronic hypoalbuminemia arises from disorders that impair albumin synthesis while simultaneously increasing catabolism or extracorporeal loss (e.g., protein-losing enteropathy, protein-losing nephropathy).
Absolute hyperalbuminemia is exceedingly rare, but has been reported in one dog and one human patient with hepatocellular carcinoma. Hyperalbuminemia was hypothesized to be a consequence of increased synthesis of albumin by malignant hepatocytes or due to decreased negative feedback from impaired hepatocellular osmoreceptivity.56,157
Globulins
The plasma globulin concentration represents many different proteins, some of which are shown in Figure 19-4. The majority of nonimmunoglobulin serum globulins are synthesized and stored in the liver. Many of these proteins function as acute-phase reactants, a group of functionally diverse proteins normally present in very small quantities. The synthesis of acute-phase proteins rapidly and markedly increases after tissue injury or inflammation under the influence of cytokines. These proteins can contribute substantially to an increased total globulin concentration. Nevertheless, determination of the total globulin concentration is not a good measure of liver synthetic function because of the contribution of immunoglobulins to the total globulin concentration.
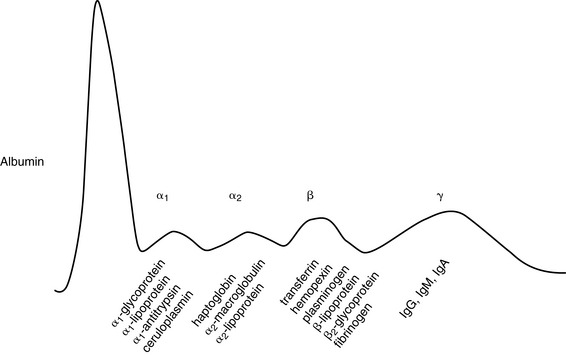
Hyperglobulinemia is common in animals with acquired hepatic disease, and the magnitude of this response may mask hypoalbuminemia if only total serum protein concentration is determined. Along with the acute-phase response, increased globulins reflect systemic immune stimulation secondary to impaired Kupffer cell function, disturbed B- and T-cell function, and development of autoantibodies. In severe hepatic insufficiency, decreased α-globulins (e.g., haptoglobin, α1-antitrypsin) and hypoalbuminemia portend a poor prognosis.190
Pathophysiology of the hepatobiliary system
Influence of liver function on blood urea nitrogen and serum creatinine
Urea Synthesis
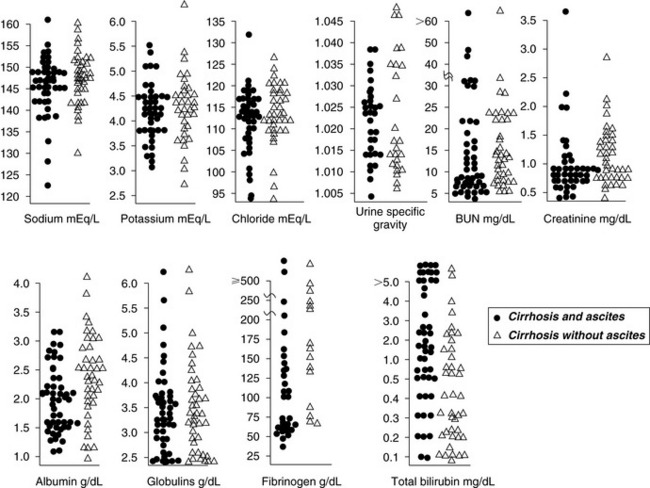
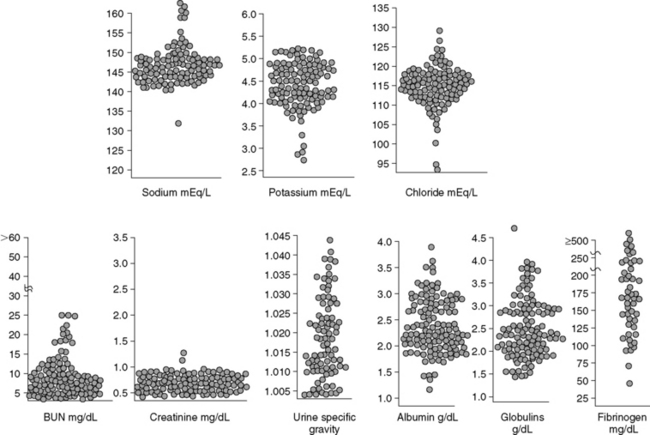
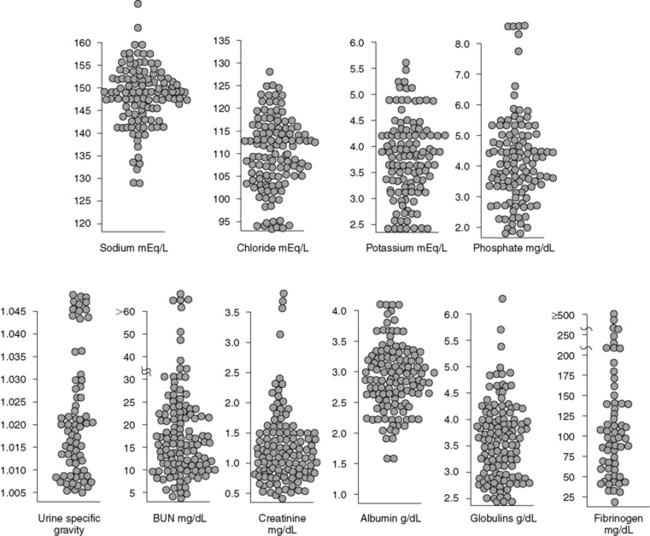
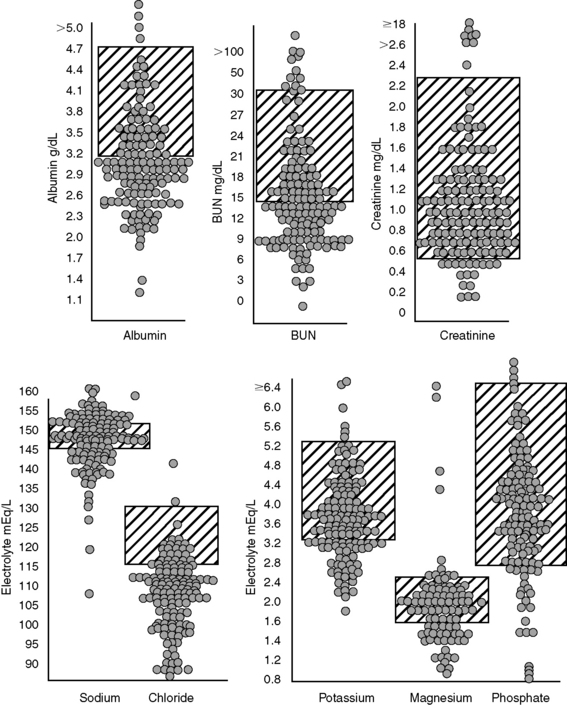
Blood urea nitrogen (BUN) concentration is directly affected by hepatic urea synthesis. Dietary protein restriction and an expanded volume of distribution for urea (e.g., hypoalbuminemia, third-space fluid accumulation, splanchnic and systemic vasodilatation) can exaggerate low BUN concentrations. Consequently, patients with acquired hepatic insufficiency and those with portosystemic shunting commonly develop abnormally low BUN concentrations. Increased water turnover associated with polydipsia and polyuria also may contribute to low BUN concentrations, whereas enteric hemorrhage in dogs with cirrhosis can increase BUN concentration into the normal range. These extrarenal factors make interpretation of BUN concentration as an indicator of renal function more difficult. BUN concentrations in dogs with cirrhosis (with and without ascites), dogs with portosystemic vascular anomaly (PSVA), and cats with hepatic lipidosis (HL) are shown in Figures 19-5, 19-6, and 19-7.
Creatinine Synthesis
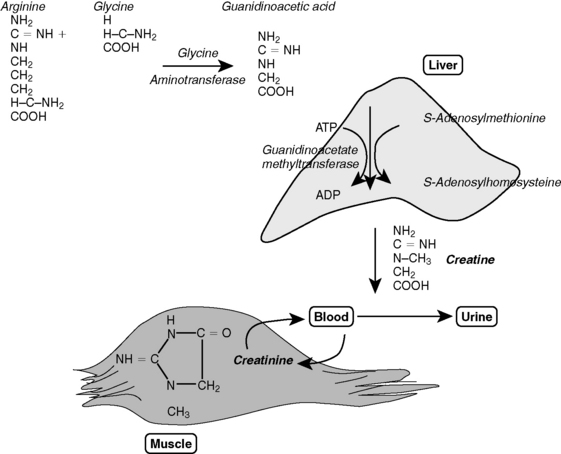
The liver also plays a major role in the biosynthesis of creatine, an organic nitrogenous compound essential for cell energy metabolism (Figure 19-8). Creatine is derived from two amino acids (arginine and lysine), and the initial synthetic step is dependent on a rate-limiting enzyme (glycine amidinotransferase) present in a wide variety of organs. The next synthetic step occurs primarily in the liver and involves the transfer of a methyl group from S-adenosylmethionine (SAMe). Decreased hepatic synthesis of creatine in liver disease can result from insufficient methylation reactions and may cause subnormal serum creatinine concentrations. Approximately 98% of creatine is located in muscle tissue. Consequently, loss of muscle mass secondary to a negative nitrogen balance (or small body size in young animals with PSVA) can cause subnormal serum creatinine concentrations (see Figures 19-5, 19-6, and 19-7). Increased water turnover associated with polydipsia and polyuria can accentuate subnormal creatinine concentrations in patients with hepatic insufficiency. In humans with hepatic cirrhosis and concurrent renal dysfunction, serum creatinine concentration fails to reflect the decreased glomerular filtration rate (GFR); a similar phenomenon may occur in animals.34,162
Hypoalbuminemia in liver disease
Hypoalbuminemia usually is accompanied by hypocalcemia (as reflected by measurement of serum total calcium concentration) as a result of decreased protein binding of calcium. It was previously thought that a linear relationship existed between serum protein and calcium concentrations in dogs and could be used to assess the clinical importance of hypocalcemia.24,144 However, total calcium concentration does not predict ionized calcium concentration in dogs.185 Therefore, it is not reliable to correct total serum calcium concentration based on serum albumin concentration.143,144 A reliable relationship between albumin, protein, and calcium concentrations also does not occur in cats.24,80
Although usually attributed to synthetic failure, hypoalbuminemia in liver disease is multifactorial. In addition to decreased synthetic capacity, increased distribution into ascites, malnutrition, and a negative acute-phase response also may affect serum albumin concentration. Increased ultrafiltration into the space of Disse (caused by sinusoidal hypertension) may overwhelm the absorptive capacity of hepatic lymphatics despite a nearly tenfold increase in lymphatic flow. Hydrostatic leakage of protein-poor ultrafiltrate from the liver aggravates abdominal effusion. In such patients, newly synthesized albumin released directly into ascitic fluid may not reach the intravascular compartment and may take weeks to equilibrate with the exchangeable albumin pool.178,243 Some human patients with severe liver disease and hypoalbuminemia maintain normal rates of albumin synthesis. In these patients, water and sodium retention are primarily responsible for hypoalbuminemia and ascites. Serum protein concentrations in dogs with hepatic cirrhosis (with and without ascites), dogs with PSVA, and cats with HL are shown in Figures 19-5, 19-6, and 19-7.
In patients with inflammatory liver disease, albumin synthesis may be suppressed by inflammatory mediators.19,38,118,152 Suppression of albumin synthesis usually is inversely proportional to the rate of acute-phase protein synthesis and thus has been called a negative acute-phase response. However, the acute-phase response also increases transcapillary diffusion of albumin. Endotoxin can increase vascular permeability to albumin, and enhanced transmural passage of endotoxins during portal hypertension may contribute to splanchnic vasodilatation and transcapillary leakage of albumin.136 Abnormal polyamine metabolism caused by altered urea cycle function and methionine metabolism also can impair albumin synthesis. Dietary restriction of protein is the most common correctable cause of hypoalbuminemia in liver disease patients. By increasing protein intake as tolerated and observing the response over weeks, the role of dietary protein restriction in hypoalbuminemia can be evaluated.
Hypoalbuminemia in liver disease generally is not accompanied by decreased globulin concentration (see Figures 19-5, 19-6, and 19-7). Rather, globulin concentration is normal or increased because of a disproportionate increase in α-globulins and acute-phase proteins. α-Globulin concentrations increase as a result of increased systemic exposure to gut-derived antigens, microorganisms, and debris normally removed by the hepatic mononuclear phagocytes (Kupffer cells) and presence of inflammatory and immune-mediated processes associated with the underlying disease. α-Globulins (particularly haptoglobin), fibrinogen, and antithrombin III are abnormally low in dogs with end-stage cirrhosis and hepatic synthetic failure.190 Portosystemic shunting and severe hepatic insufficiency also decrease plasma concentration of protein C, an important anticoagulant also involved in the inflammatory response.222,223 The diagnostic utility of the serum total protein concentration is complicated by the induction of haptoglobin by glucocorticoids and development of coagulopathies that can further deplete fibrinogen, antithrombin III, and protein C.98,112
The wide range of serum albumin concentrations in normally hydrated cirrhotic dogs with and without ascites demonstrates that hypoalbuminemia is only one factor influencing ascites formation (see Figure 19-5). In dogs with ascites (n = 52), median serum albumin concentration was 2.0 g/dL (range, 1.2 to 3.2 g/dL), and in dogs without ascites (n = 50), median serum albumin concentration was 2.4 g/dL (range, 0.7 to 4.2 g/dL). Median serum globulin concentrations in these dogs were similar, whereas median plasma fibrinogen concentration was significantly decreased in ascitic dogs (median, 105 mg/dL; range, 30 to 780 mg/dL) compared with dogs without ascites (median, 165 mg/dL; range, 64 to 550 mg/dL).
Serum electrolytes
Hypokalemia in Liver Disease
Hypokalemia is a serious electrolyte disturbance associated with hepatic insufficiency.37 Contributing factors include insufficient energy intake, enteric losses (e.g., vomiting, diarrhea, nutrient malassimilation), treatment with loop diuretics, and secondary hyperaldosteronism.30,213,221 Magnesium deficiency also can complicate hypokalemia by potentiating kaliuresis through its effects on aldosterone.84 Hypokalemia may go unrecognized because of the transcellular shift that occurs between potassium and hydrogen ions. Serum potassium concentrations of dogs with cirrhosis, dogs with PSVA, and cats with HL are shown in Figures 19-5, 19-6, and 19-7. Frank hypokalemia was present in 11 of 48 cirrhotic dogs with ascites, in 10 of 42 of cirrhotic dogs without ascites, in 6 of 113 dogs with PSVA, and in 32 of 116 cats with HL. A total of 34 of 90 cirrhotic dogs (19 of 48 with ascites and 15 of 42 without ascites), 24 of 104 dogs with PSVA, and 44 of 116 cats with HL had subnormal or low normal serum potassium concentrations. Although the prognosis is worse for cats with HL and hypokalemia, the prognostic significance of hypokalemia has not been evaluated in the other disorders.39
It is important to recognize and correct hypokalemia for several reasons. Most importantly, a reciprocal relationship exists between intracellular and extracellular potassium concentrations and renal ammoniagenesis.93,212,213 Infusion of potassium chloride in hypokalemic patients significantly improved central nervous system (CNS) function in early hepatic encephalopathy (HE) and prolonged survival in cirrhotic humans.242 Patients given potassium chloride to establish normokalemia experienced decreased arterial NH3 concentration and pH, increased arterial 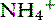 /NH3 ratio, decreased urine pH, and slightly increased 24-hour urinary ammonia excretion with a significantly increased urine
/NH3 ratio, decreased urine pH, and slightly increased 24-hour urinary ammonia excretion with a significantly increased urine 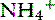 /NH3 ratio. Mechanistically, potassium infused into the hypokalemic patient replaces intracellular hydrogen ions. The displaced cellular hydrogen ions decrease blood pH, promoting conversion of NH3 to the less-diffusible
/NH3 ratio. Mechanistically, potassium infused into the hypokalemic patient replaces intracellular hydrogen ions. The displaced cellular hydrogen ions decrease blood pH, promoting conversion of NH3 to the less-diffusible 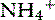 form. This small shift in pH is not great enough to stimulate renal ammoniagenesis, but reduced urine pH leads to increased excretion of
form. This small shift in pH is not great enough to stimulate renal ammoniagenesis, but reduced urine pH leads to increased excretion of 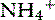 . This effect may be augmented by increased plasma aldosterone given its ability to increase hydrogen ion delivery into distal renal tubular fluid.188
. This effect may be augmented by increased plasma aldosterone given its ability to increase hydrogen ion delivery into distal renal tubular fluid.188
Serum Potassium Concentration and Ammoniagenesis
Experimental and clinical observations of potassium depletion and loading suggest that renal NH3 production is intimately linked with potassium homeostasis. Low serum potassium concentrations stimulate and high serum potassium concentrations suppress renal ammoniagenesis.154,214 A closed-loop regulatory system modulates NH3 production, hydrogen ion homeostasis, and urinary potassium excretion in response to acute and chronic changes in serum potassium concentration. Potassium deficiency stimulates H+ secretion in the distal nephron and may stimulate 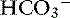 production by increasing collecting duct expression of an H+-K+-ATPase that facilitates reabsorption of K+ in exchange for H+.119,154 Potassium deficiency also may increase luminal electronegativity in the proximal tubule, stimulating
production by increasing collecting duct expression of an H+-K+-ATPase that facilitates reabsorption of K+ in exchange for H+.119,154 Potassium deficiency also may increase luminal electronegativity in the proximal tubule, stimulating 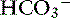 secretion.31 Hypokalemia arising from diuretics used to treat ascites can cause hyperammonemia secondary to metabolic alkalosis resulting from renal H+ loss.
secretion.31 Hypokalemia arising from diuretics used to treat ascites can cause hyperammonemia secondary to metabolic alkalosis resulting from renal H+ loss.
Hypophosphatemia in Liver Disease
Hypophosphatemia also may complicate hepatic insufficiency. In human patients, hypophosphatemia and early phosphorus administration are associated with a good prognosis in acute liver failure, whereas hyperphosphatemia is predictive of poor recovery.18 Cats with HL are at increased risk for development of hypophosphatemia, especially when associated with diabetes mellitus or pancreatitis. Although symptomatic hypophosphatemia may develop after rehydration and insulin therapy, it is most common as a result of refeeding in cats with HL.6 Serum potassium, magnesium, and phosphorus concentrations in 157 cats with severe HL are shown in Figure 19-9. In this population, only 22 of 157 (14%) HL cats had hypophosphatemia at presentation, but more than 35% of those undergoing nutritional support became hypophosphatemic with refeeding. Hypophosphatemia in patients with liver disease is thought to reflect intracellular shifts of phosphate.81,206 Although less common on presentation than hypokalemia, severe hypophosphatemia can produce many clinical signs including weakness (e.g., ventilatory failure severe enough to cause respiratory acidosis, neck ventroflexion in cats), vomiting, gastric atony, hemolysis, bleeding tendencies (i.e., platelet dysfunction), hemolytic anemia, and neurologic signs that can be confused with HE.59,81 Mechanisms of hemolysis involve depletion of red cell energy related to impaired glycolysis and ATP production and diminished ability to maintain reduced GSH in erythrocytes. Muscle weakness in hypophosphatemia may be severe enough to impair ventilation, leading to ventilatory failure and respiratory acidosis. Hypophosphatemia induced by refeeding in cats with HL typically appears within the first 48 hours of alimentation, and overt clinical effects are observed with serum phosphorus concentrations less than 1.5 mg/dL.
Hypomagnesemia in Liver Disease
Symptomatic hypomagnesemia is observed infrequently in patients with liver disease. Recognition of low serum magnesium concentration is important because of the essential role of magnesium as an enzyme cofactor. The mechanisms underlying clinical signs have not been clarified but likely involve transcellular shifting of magnesium into cells with glucose. Hypomagnesemia also may be induced by citrate toxicity after large-volume transfusion with citrate-phosphate-dextrose (CPD)-anticoagulated blood in patients with limited ability for hepatic metabolism of citrate. The most important clinical manifestations of hypomagnesemia are muscle weakness, impaired contractility of the diaphragm, aggravation of preexisting cardiomyopathy, and altered sensorium that may mimic HE. These clinical signs also can be mistakenly attributed to abnormal serum potassium or phosphorus concentrations. Additionally, severe hypomagnesemia can impair the response to potassium supplementation because it perpetuates renal potassium wasting.46
Water and sodium disturbances in chronic liver disease
Iso-osmotic Renal Sodium Retention
In many patients with hepatic insufficiency prone to ascites formation, iso-osmotic renal sodium retention expands extracellular volume such that total body sodium is not reflected in the serum sodium concentrations (see Figure 19-5). In humans, the magnitude of sodium retention varies among individuals. Hyponatremia in critically ill cirrhotic patients is associated with a poor short-term prognosis. Serum sodium concentration is an important predictor of survival among candidates for liver transplantation.17,19–22 Serum sodium concentrations less than 123 to 135 mEq/L have been associated with a poor outcome.25,35,86,108,113 In one study, however, low serum sodium concentration was found to reflect poor renal function, and did not affect survival when corrected for the GFR.129 Sodium retention also varies in cirrhotic dogs and is indicated by their diverse urine specific gravity (USG) values and serum sodium concentrations at presentation and their apparent resistance to diuretic therapy.
Impaired Excretion of Solute-Free Water
Up to 35% of human patients with cirrhosis develop impaired free water excretion causing dilutional hyponatremia.163,165,193 A similar phenomenon may occur in dogs (see Figure 19-5).17,73,140,164,227 When dogs with cirrhosis with and without ascites were compared, the overall frequency of hyponatremia on initial presentation was approximately 25% with the lowest serum sodium concentrations found in dogs with ascites (see Figure 19-5). In humans, serum sodium concentrations of 130 mEq/L corresponded with higher risk of ascites, hepatic encephalopathy, bacterial peritonitis, and hydrothorax, compared with the risks in patients with serum sodium concentration of 136 mEq/L. However, serum sodium concentration has not been associated with the presence of varices.113 In dogs, marked hyponatremia was only observed in association with substantial free water retention and ascites.
Pathophysiology of Fluid Retention in Cirrhosis
In cirrhosis, disturbances in fluid balance precede ascites formation by several weeks. In this phase, intravascular volume expansion results from renal sodium retention.140 Renal tubular sodium retention also precedes changes in renal blood flow, GFR, filtration fraction, and intrarenal vascular resistance associated with cirrhosis.127 A 36% plasma volume expansion occurred in cirrhotic dogs during this active salt-retaining, preascitic phase, with two thirds of the newly acquired volume distributed to the vasodilated splanchnic circulation.126 Ascites formation is hastened by sodium ingestion or intravenous administration of sodium-containing fluids. Surgical creation of portosystemic shunting in dogs with hepatic cirrhosis abolished portal hypertension and the early tendency for renal sodium retention and ascites. In such studies, 20- to 30-lb cirrhotic dogs with shunts were able to maintain normal sodium balance with intakes as high as 85 mEq/day. Cirrhotic dogs without shunts accumulated sodium at this level of intake.225
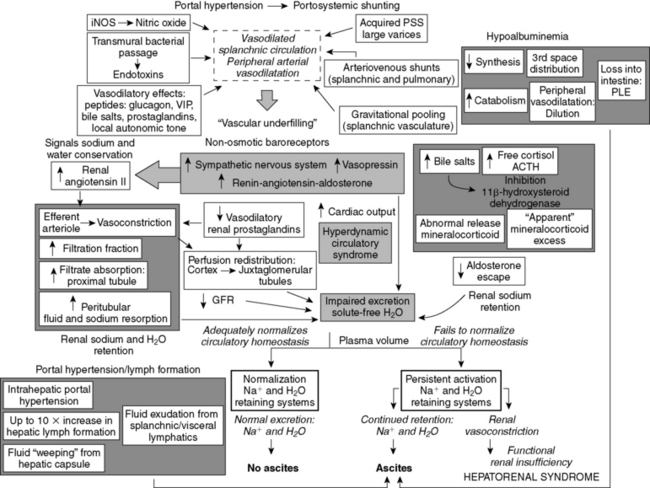
Peripheral arterial and splanchnic vasodilatation initiates water and sodium conservation in cirrhosis.90 Peripheral arterial vasodilatation (“underfilling”) reenforces the signal initiating renal sodium retention (i.e., perceived reduction in circulating ECF volume). The physiologic responses observed after acute portal vein constriction (i.e., systemic arterial vasodilatation and hypotension, ECF expansion, increased cardiac output) are similar to those associated with the hyperdynamic circulatory syndrome of cirrhosis.21
These hemodynamic maladjustments are mediated by the renin-angiotensin-aldosterone system (RAAS) and SNS in response to underfilling of the systemic arterial circulation and decreased renal perfusion.25,86 Abnormal intrarenal accumulation of angiotensin II occurs early in the disease process, even before activation of the RAAS.125 Renal sodium conservation may be related in part to enhanced sensitivity to aldosterone.
Effect of Portosystemic Shunting on Sodium and Water Retention
Portosystemic shunting also may affect sodium and water retention, and surgically created portosystemic shunts in experimental dogs have been used to study the effects of diverted hepatoportal perfusion on sodium and water balance. Ten weeks after end-to-side portocaval shunt formation, plasma volume, systemic blood pressure, and central venous pressures were maintained, and no changes in GFR, plasma renin activity, or aldosterone concentrations were identified.124 Some dogs maintained normal sodium balance after ingestion of 150 mEq/day of sodium, but others developed ascites.124 These findings indicate that in some situations portosystemic shunting alone can impair ability to adapt to increased sodium loads. This finding may explain the tendency to form ascites in some dogs with PSVA (especially those with ductus venosus) and hypoalbuminemia or after administration of sodium-rich crystalloids.
Specific Mechanisms of Water and Electrolyte Disturbances in Cirrhosis and Portosystemic Shunting
Nonosmotic Vasopressin Stimulation
Nonosmotic stimulation of AVP is a central factor mediating water retention in cirrhosis.90 Acute changes in portal venous pressure in cirrhotic dogs initiate AVP-mediated antidiuresis. Both systemic and splanchnic arterial vasodilatation can stimulate nonosmotic AVP release and activate other antidiuretic and vasopressor systems.86,90 Early in cirrhosis (“compensated cirrhosis”), transient neurohormonal responses increase plasma volume and temporarily suppress baroreceptor signaling. As the disease progresses, arterial vasodilatation worsens, and neurohormonal responses are no longer able to compensate. At this point, vasoconstrictor systems become continuously stimulated and promote the sodium and water retention that causes edema and ascites. The response is exaggerated by abnormal retention of AVP as a result of impaired metabolism. Normally, the kidney and liver metabolize AVP, but decreased AVP clearance in hepatic disease correlates with disease severity.200 Conivaptan, a nonpeptide, dual V1a/V2 AVP receptor antagonist has shown promising results in both animals240 and humans.9 It binds competitively and reversibly with high affinity to the V1a and V2 receptors that mediate vasoconstriction and water permeability, respectively. Conivaptan has been shown to correct hyponatremia in euvolemic or hypervolemic patients.9
Increased Basal Cortisol and ACTH Concentrations
Increased basal cortisol and adrenocorticotropic hormone (ACTH) concentrations complicate hepatic insufficiency associated with acquired portosystemic shunting in dogs, but normal adrenal response to low-dose dexamethasone suppression is maintained.180 High basal cortisol concentrations also were found in dogs with congenital PSVA, and concentrations normalized after successful shunt ligation.203 In another report, baseline cortisol concentrations in dogs with congenital PSVA and in healthy dogs undergoing ovariohysterectomy were similar. Response to ACTH did not correlate with postoperative hypoglycemia or prolonged anesthetic recovery, which was previously thought to be due to inadequate adrenal respose.105 Dogs with PSVA also have high free-water flux and an abnormally high GFR that normalize after shunt ligation.65 It is unknown if this response relates to abnormal cortisol concentration or hemodynamic adjustments. Other potential causes for hypercortisolemia in dogs with congenital PSVA include decreased hepatic synthesis of cortisol binding proteins, decreased hepatic clearance of cortisol, peripheral resistance to cortisol, or stress associated with chronic nonadrenal illness.105
Altered Steroid Hormone Metabolism
Altered steroid hormone metabolism also may contribute to sodium retention in cirrhosis. Abnormally increased serum bile salt concentrations may inhibit 11β-hydroxysteroid dehydrogenase-2 (11β-HSD-2), the enzyme that interconverts endogenous and exogenous biologically active 11β-hydroxysteroids and their inactive 11-ketosteroid counterparts. 11β-HSD-2 selectively modulates access of aldosterone to mineralocorticoid receptors and normally is located in mineralocorticoid-responsive tissues (including the distal nephron). Absence or inhibition of 11β-HSD-2 can mimic mineralocorticoid excess by allowing inappropriate access of 11β-hydroxyglucocorticoids to mineralocorticoid receptors.4,114,171 The up-regulation of the vasopressin-regulated water channel aquaporin-2 (AQP2) and increased targeting of AQP2 to luminal membranes likely to contribute to the increased water reabsorption and urinary concentration in hepatic cirrhosis.114
Abnormal Aldosterone Release and Responsiveness to Aldosterone
High (or inappropriately normal) aldosterone concentrations precede and accompany pathologic sodium retention in humans and animals with cirrhosis. Experimentally, hepatic venous congestion and acute portal hypertension stimulate aldosterone secretion.23 The importance of aldosterone in sodium and water retention in cirrhosis in humans is demonstrated by the efficacy of spironolactone (a specific aldosterone antagonist) in mobilizing ascites and alleviating sodium retention in patients without underlying renal dysfunction. The influence of aldosterone on renal sodium retention is enhanced by increased renal sensitivity to the hormone. This phenomenon is reflected clinically by decompensation (i.e., ascites induction) of cirrhotic dogs given glucocorticoids with minimal mineralocorticoid activity (e.g., prednisone).
Splanchnic Arterial Vasodilatation
Although the cause of systemic and splanchnic arterial vasodilatation that stimulates AVP production and other antidiuretic and vasopressor mechanisms is not completely understood, nitric oxide (NO) plays an integral role. Splanchnic NO is produced by inducible NO synthetase activity in the mesenteric splanchnic endothelium. Splanchnic vasodilatation also reflects formation of arteriovenous shunts, acquired portosystemic communications, and other endothelial (e.g., prostacyclin, endothelin) and nonendothelial (e.g., glucagon, vasoactive intestinal peptide) vasodilatory mechanisms.11 Vasodilatation of splanchnic vasculature also may reflect increased exposure to bacterial endotoxins from enhanced transmural passage of endotoxin from the gut lumen.208
Diminished Renal Prostaglandin Synthesis
Decreased renal prostaglandin production increases pathologic water accumulation and dilutional hyponatremia in cirrhosis and hepatorenal syndrome (HRS [see the Hepatorenal Syndrome section]).90 Endogenous renal prostaglandins normally play an important role in regulation of renal perfusion and tubular response to AVP, especially when vasoconstrictor forces predominate (as in cirrhosis). Renal synthesis of vasodilatory eicosanoids (e.g., prostaglandin [PG] I2 and PGE2) normally counterbalances vasoconstrictive stimuli (e.g., angiotensin II, AVP, increased renal sympathetic tone) and preserves renal blood flow and GFR. The protective effect of renal prostaglandins becomes apparent when cirrhotic patients with ascites are treated with nonsteroidal antiinflammatory drugs (NSAIDs). These patients may experience decreased renal blood flow and GFR, activation of vasoconstrictor systems, and sodium and fluid retention that can cause acute renal failure and HRS.
Water and Sodium Disturbances in Cats with Liver Disease
Cats with HL do not have consistent changes in serum electrolyte concentrations (see Figure 19-7). This finding is not unexpected because many conditions that cause anorexia and rapid weight loss lead to HL. In a survey of cats with severe HL, 14 of 72 had USG values less than 1.010, 29 of 114 were hyponatremic, and only 1 was hypernatremic. Cats with chronic cholangitis or cholangiohepatitis also do not have consistent changes in serum sodium concentration or USG.
Summary of Effects of Cirrhosis on Total Body Sodium and Water and Ascites Formation
In cirrhotic patients, there is a relative inability to adjust water excretion to the amount of water ingested and decreased ability to eliminate sodium in the urine. Impaired water and sodium elimination arises from several factors: (1) enhanced sodium reabsorption in the proximal nephron and decreased delivery of glomerular filtrate to the distal nephron; (2) decreased GFR caused by splanchnic vasodilatation, low systemic blood pressure, altered cardiac output, and inappropriate vasoconstriction of the glomerular efferent arterioles; (3) decreased renal prostaglandin synthesis (PGE2) and impaired autoregulation of renal blood flow; (4) pathologic redistribution of renal blood flow away from the cortex; (5) increased response to, or activity of, aldosterone; and (6) nonosmotic stimulation of AVP release. The most important factors favoring dilutional hyponatremia are disturbed hemodynamics involving the splanchnic and systemic circulation and nonosmotic AVP release. Medical treatment of impaired water and sodium is difficult and may be facilitated by aquaretic agents and vasopressors specific for the splanchnic circulation.89,116,235 In the future, some patients may benefit from treatment with conivaptan to antagonize the effects of AVP.240 The importance of sodium retention in ECF volume expansion associated with portal hypertension is evidenced by patient response to dietary sodium restriction and diuretic stimulation of natriuresis. The severity of sodium retention relative to water retention varies among individuals, and serum sodium concentration does not predict ascites formation (see Figure 19-5). Some patients produce urine that is virtually free of sodium, whereas others produce inappropriately concentrated urine because of excessive AVP release and are at high risk for dilutional hyponatremia.
Ascites resulting from liver disease
Pathophysiologic mechanisms underlying ascites formation are complex, and no specific clinical features clearly identify patients prone to ascites formation. Serum electrolyte, BUN, creatinine, protein, and total bilirubin concentrations for 109 cirrhotic dogs with and without ascites are shown in Figure 19-5. Better understanding of the pathophysiology of ascites formation has led to a shift from the classical underfilling and overflow hypotheses to the forward theory (Figure 19-10). Currently, splanchnic arterial vasodilatation and associated systemic and renal counter-regulatory responses are thought to be the main pathophysiologic events underlying ascites formation. Decreased systemic vascular resistance initially arises as a consequence of marked splanchnic arterial vasodilatation. The mechanisms underlying splanchnic vasodilatation are poorly understood but likely involve enhanced availability, synthesis, or activity of vasodilatory factors such as NO, glucagon, vasoactive intestinal peptide, endotoxin, bile acids, prostaglandins, and increased local autonomic tone. Splanchnic vasodilatation promotes abnormal distribution of circulating blood volume away from the systemic circulation. The resulting systemic hypoperfusion is sensed by arterial baroreceptors, which signal a need for vasoconstriction and sodium and water retention by the kidneys (e.g., activation of the RAAS and SNS, release of AVP). These events establish a hyperdynamic state characterized by increased cardiac output, decreased systemic vascular resistance, and arterial vasodilatation affecting both the splanchnic and systemic circulation.
Albumin infusions do not consistently improve circulatory and renal function in cirrhotic patients with ascites because of enhanced movement of albumin from vessels into the interstitium and severe vasodilatation of the splanchnic circulation. Although acute volume expansion in cirrhotic human patients increases peripheral blood volume, limited improvement occurs in central blood volume (i.e., splanchnic, hepatic, and cardiopulmonary circulation). However, infusion of albumin in combination with administration of terlipressin, a long-acting synthetic AVP analog, can cause splanchnic vasoconstriction and improved systemic perfusion.226
Assessment of Ascites
A sample of the abdominal effusion should be evaluated biochemically, cytologically, and by culture if cytology suggests infection. Ascites arising from liver disease typically is a pure transudate with a total protein concentration of less than 2.5 g/dL and a specific gravity between 1.010 and 1.015. Cytologically, the fluid has low cellularity with only a few mesothelial cells and neutrophils present. In the jaundiced patient, the fluid is yellow and bilirubin crystals may be observed, but the bilirubin concentration of the effusion is less than that of serum. A serum-to-effusion albumin gradient greater than 1.1 suggests portal hypertension as a causative mechanism.170 Body weight and abdominal girth measurements should be taken as a reference for evaluating changes in fluid accumulation. Girth measurements are meaningful only if a consistent method is used. A mark is made on the abdomen with a permanent ink pen, and the owner is taught to monitor ascites accumulation by measuring girth circumference using a consistent technique.
Abnormal renal function in liver disease
As liver function deteriorates and portal hypertension worsens, several maladaptive responses threaten renal function. Decreased GFR reduces delivery of glomerular filtrate to the distal diluting segments of the nephron. Coupled with increased resorption in the proximal tubule, this increases renal sodium and water reabsorption, impairs renal escape from abnormally increased aldosterone, and favors resistance to atrial natriuretic peptides.72 Systemic counter-regulatory responses that normally preserve filtration fraction increase production of angiotensin II and further provoke vasoconstriction of the efferent arterioles. Although these events maintain glomerular capillary pressure, increase filtration fraction, and alter peritubular Starling’s forces favoring fluid reabsorption, they do so at the expense of decreased renal blood flow.90 Functional disruption of solute conservation in Henle’s loop by loop diuretics (e.g., furosemide) may further impair the ability of the nephron to dilute or concentrate urine.
Increased Water Turnover and Glomerular Filtration Rate
The influence of hepatic insufficiency on BUN and serum creatinine concentrations is aggravated by increased water turnover and development of a supranormal GFR as observed in dogs with PSVA.65 Primary polydipsia associated with HE, stimulation of hepatoportal osmoreceptors, and an impaired renal medullary concentration gradient (e.g., chronic hypokalemia, decreased urea synthesis) may contribute to abnormal water balance in these animals.95,120,214
Polyuria and Polydipsia
Polydipsia, polyuria, and renal dysfunction may be associated with liver disease in both dogs and cats. Dogs with PSVA may be presented primarily for evaluation of polyuria and polydipsia.41,95 Mechanisms may include psychogenic polydipsia associated with HE; sensory input signaling splanchnic vasodilatation, decreased hepatic portal perfusion, or altered osmolality; renal medullary washout caused by low urea concentration; renal tubular dysfunction associated with potassium depletion; or increased concentrations of endogenous steroids.120
Evaluation of USG before fluid therapy in dogs with PSVA showed that 47 of 87 had a USG less than 1.020, and 12 of 87 were hyposthenuric (see Figure 19-6). Serum electrolyte concentrations were not significantly correlated with USG, but subnormal BUN concentrations occurred in 58 of 123 dogs, and low normal or subnormal creatinine concentrations were found in 83 of 123. These findings suggest that diuresis contributes to low USG in these patients, as supported by presence of a supranormal GFR in dogs with PSVA.65 Subnormal BUN concentrations in dogs with PSVA could impair maintenance of the renal medullary solute gradient necessary for water reabsorption in response to AVP. Low serum creatinine concentration probably reflects reduced muscle mass associated with the young age and small size of many affected dogs, hepatic insufficiency, and increased water turnover.34,65,103,162
Similar mechanisms are likely to be operative in dogs with acquired hepatic insufficiency. Of cirrhotic dogs with ascites, 15 of 26 with urinalysis performed before treatment had a USG less than 1.020 (see Figure 19-5). Of these, only 3 of 26 were hyposthenuric. In the same group. 11 of 42 had low BUN concentrations, and 21 of 42 had low or subnormal serum creatinine concentrations. In cirrhotic dogs without ascites, 16 of 34 with urinalysis performed before treatment had a specific gravity less than 1.020, and only 1 of 34 was hyposthenuric. In the same group. 20 of 47 had low BUN concentrations, and 36 of 47 had low normal or subnormal serum creatinine concentrations.
Altered Intrarenal Hemodynamics
Subtle changes in intrarenal hemodynamics contribute to deranged renal function in cirrhosis. Normally, renal blood flow is predominantly distributed to cortex (90%) with less blood flow to the outer (9%) and inner medulla (1%). Autoregulation of renal blood flow maintains proper balance between afferent and efferent arteriolar tone to regulate the GFR and filtration fraction. Redistribution of blood flow from the outer cortical to juxtamedullary nephrons occurs in approximately 60% of human patients with ascites. Redistribution of renal blood flow and increased intrarenal arterial resistance are correlated with increased plasma renin activity.21,117 Changes in systemic and splanchnic hemodynamics (e.g., low systemic arterial blood pressure, decreased systemic vascular resistance, splanchnic vasodilatation) associated with the hyperdynamic circulatory state of cirrhosis initiate renal vasoconstrictor responses that further compromise renal perfusion. Arterial vasodilatation expands vascular capacity and makes effective circulating blood volume difficult or impossible to maintain. High SNS activity further reduces renal cortical blood flow, whereas low systemic pressure and increased renal interstitial pressure compromise renal blood flow, GFR, sodium excretion, and water diuresis.
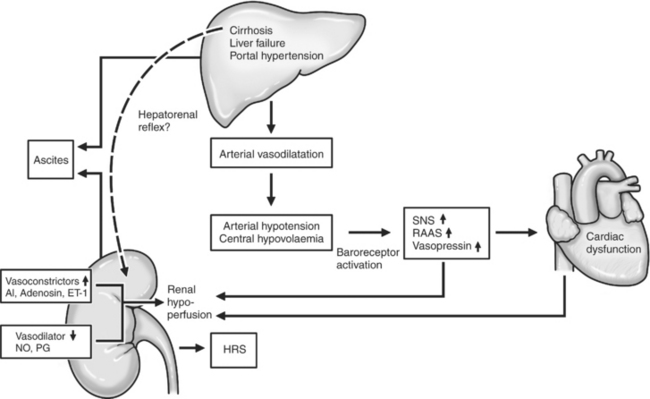
Hepatorenal Syndrome
HRS is a state of functional renal failure associated with a low GFR, preserved tubular function, and normal renal histology that occurs in some human patients with cirrhosis and ascites.147 A similar syndrome rarely may occur in veterinary patients. Reduced renal cortical perfusion resulting from increased renal vascular resistance precedes renal failure in this syndrome. The cause of intrarenal vasoconstriction is complex and poorly understood (Figure 19-11). Factors associated with development of HRS in humans are listed in Box 19-1. Essential diagnostic criteria for HRS in humans include a spontaneously acquired acute decline in the GFR, impaired urinary sodium excretion (<10 mEq/day), urine osmolality greater than plasma osmolality, and the absence of other causes of renal failure.
Prevention of HRS requires early intervention to minimize circulatory instability and renal hypoperfusion. Treatment in human patients has included plasma expanders (e.g., albumin, colloids), the long-acting α-adrenergic agonist midodrine to improve systemic blood pressure and renal perfusion, and the somatostatin analog octreotide and the AVP analog terlipressin to attenuate splanchnic vasodilatation.3,12,116,183,226 In the future, endothelins, adenosine antagonists, long-acting vasoconstrictors, and antileukotriene drugs may play a role in preventing and treating HRS.146
Acid-base disturbances in liver disease
Although experimental studies support a role for hepatic urea and glutamine cycles in regulation of systemic pH by their effects on renal ammoniagenesis, there is no consistent pattern of acid-base disturbances in patients with liver disease.150,191 The most common disturbance in humans with hepatic insufficiency and coma is respiratory alkalosis, but metabolic acid-base disturbances may also occur.150,173,191 Patients with stable cirrhosis and those with portal hypertension attenuated by surgically created portosystemic shunts commonly develop compensated respiratory or metabolic alkalosis. Respiratory alkalosis is closely associated with the extent of functional liver impairment rather than the presence of portosystemic shunting and nearly always is compensated.173
Mechanism of Respiratory Alkalosis
Respiratory alkalosis in cirrhosis may evolve subsequent to reduced arterial oxygen saturation secondary to acquired venoarterial shunting, ventilation-perfusion mismatch (derived from ascites-induced restriction of ventilatory efforts or changes in pulmonary capillaries), a shift to the right in the oxyhemoglobin dissociation curve, direct stimulation of the respiratory center by encephalopathic toxins (e.g., NH3), or development of CNS acidosis.101 Respiratory alkalosis may also develop as compensation for metabolic acidemia (e.g., lactic acidosis, increased concentrations of free fatty acids, impaired renal tubular acid excretion, or renal hypoperfusion).10,176
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


