Chapter 7 Disorders of Phosphorus
Hypophosphatemia and Hyperphosphatemia
Phosphorus plays an essential role in cellular structure and function.91 A constituent of structural phospholipids in cell membranes and hydroxyapatite in bone, phosphorus also is an integral component of nucleic acids and phosphoproteins involved in mitochondrial oxidative phosphorylation. Energy for essential metabolic processes (e.g., muscle contraction, neuronal impulse conduction, epithelial transport) is stored in high-energy phosphate bonds of adenosine triphosphate (ATP). The compound 2,3-diphosphoglycerate (2,3-DPG) decreases the affinity of hemoglobin for oxygen and facilitates the delivery of oxygen to tissues. Cyclic adenosine monophosphate (cAMP) is an intracellular second messenger for many polypeptide hormones. Phosphate is also an important urinary buffer, and urinary phosphate constitutes the majority of titratable acidity (see Chapter 9).
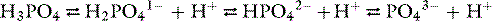
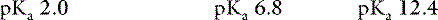
Body stores and distribution
Phosphate is the body’s major intracellular anion, and translocation in and out of the intracellular compartment can rapidly change serum phosphorus concentration. Gradual changes in total body phosphate can be accommodated without noticeable changes in serum phosphorus concentration, resembling the situation with potassium (the major intracellular cation). Approximately 80% to 85% of total body phosphate is inorganic hydroxyapatite in bone, whereas 15% is in soft tissues such as muscle.59,88 Most soft tissue phosphorus is organic and can be readily converted to the inorganic form as needed. The ECF compartment contains less than 1% of total body phosphorus stores.
Normal serum concentrations
Normal serum phosphorus concentrations in adult dogs range from 2.5 to 6.0 mg/dL, but they are higher in dogs younger than 1 year.17,77,131,175 Serum phosphorus concentrations are highest in puppies less than 8 weeks of age (up to 10.8 mg/dL may be considered normal) and gradually decrease into the adult range after 1 year of age.73 Sex-related changes are not reported.134 The effect of age is less pronounced in cats, but immature cats have a tendency for higher serum concentrations.32 Bone growth and an increase in renal tubular reabsorption of phosphorus mediated by growth hormone presumably contribute to this age effect. Feeding also affects serum phosphorus concentration. A carbohydrate meal or infusion (e.g., 5% dextrose) decreases serum phosphorus concentration, because phosphate shifts into intracellular fluid as a result of stimulation of glycolysis and formation of phosphorylated glycolytic intermediates in muscle, liver, and adipose cells. In contrast, protein intake increases serum phosphorus concentration because of the relatively high phosphorus content of protein-rich diets.
Time of sampling affects the observed serum phosphorus concentration. People have substantial variations in serum phosphorus concentrations throughout the day.99 Acid-base balance also influences serum phosphorus concentration. Respiratory alkalosis stimulates glycolysis (by activating phosphofructokinase) and decreases serum phosphorus concentration. Thus, the measured serum phosphorus concentration is affected by several variables and does not accurately indicate total body phosphorus stores. Measuring serum phosphorus concentration after a 12-hour fast minimizes confounding factors, but the clinician must understand that the magnitude of hypophosphatemia or hyperphosphatemia may be incorrectly assessed if only one serum or plasma sample is analyzed.
Hemolysis may affect laboratory results because phosphate is present in erythrocytes. Human erythrocytes contain 8 μmol/dL red cells, whereas canine erythrocytes contain 35 μmol/dL and feline erythrocytes contain 26 μmol/dL.30 Hyperlipidemia and hyperproteinemia sometimes cause overestimation of serum phosphorus concentration, depending on the methodology used.27,68,97 This can become important when using drugs such as liposomal amphotericin B.95 Thrombocytosis and monoclonal gammopathy also may cause spurious increases in serum phosphorus concentration.92,103,108 Mannitol and other drugs may interfere with some assay systems, leading to erroneous measured values.62,179 Icterus and hemolysis were reported to result in artifactual hypophosphatemia in dogs with immune-mediated hemolytic anemia.72 Artifactual hypophosphatemia can occur in some automated systems but not in others. Thus, occurrence of hypophosphatemia in patients without known predisposing factors should prompt consideration of laboratory error.
Dietary intake
The average phosphorus content of commercial pet foods is approximately 1% on a dry matter basis. Dogs and cats need to ingest 0.5 to 3.0 g of phosphorus per day, depending on their body size and energy requirements. The source of dietary phosphorus markedly affects absorption and excretion of phosphorus in cats.56 The amount of phosphorus absorbed by the gastrointestinal tract, the amount excreted in the urine, and the extent of postprandial hyperphosphatemia were increased when monobasic and dibasic salts of phosphorus were fed but decreased when phosphorus originated from poultry, meat, and fish meal.
Intestinal absorption
There is no evidence of a direct effect of parathyroid hormone (PTH) on intestinal phosphate absorption, and observed effects are probably mediated by the role of PTH in conversion of 25-hydroxycholecalciferol to calcitriol. High dietary ratios of calcium to phosphorus (>3 to 4) may suppress intestinal phosphate absorption, presumably through binding of phosphate by calcium and formation of poorly absorbed calcium phosphate complexes. During phosphate deprivation, the kidneys dramatically reduce phosphate excretion to negligible amounts in fewer than 3 days. Obligatory gastrointestinal loss continues for at least 3 weeks, but there is a diminution in the amount lost.99 This gastrointestinal loss may cause a cumulative negative phosphorus balance during phosphate deprivation.
Renal handling
The kidneys adjust tubular reabsorption of filtered phosphate to maintain zero balance. Normally, 80% to 90% of the filtered phosphate load is reabsorbed by the renal tubules, and renal dysfunction is the most common cause of hyperphosphatemia beside that found in young dogs.31,160
Phosphate crosses the luminal membranes of the proximal renal tubular cells by brush border sodium-phosphate cotransporters. The main transport protein in the proximal tubules (type IIa sodium-phosphate cotransporter) translocates three sodium ions and one divalent phosphate ion across the luminal membrane and thus promotes luminal electronegativity.168 Luminal entry is the rate-limiting step and the target for physiologic and pathophysiologic mechanisms that alter phosphate reabsorption.115 High dietary intake of phosphorus decreases proximal tubular reabsorption, whereas low dietary intake can result in nearly 100% proximal tubular reabsorption of phosphate. These dietary effects occur independently of changes in the plasma concentrations of phosphaturic hormones. PTH is the most important regulator of renal phosphate transport, and it decreases the tubular transport maximum for phosphate reabsorption (TmaxPi) in the proximal tubule where most phosphate reabsorption occurs. Apparently, no reabsorption occurs in the thin ascending limb or thick ascending limb of Henle’s loop, and the presence of a reabsorptive mechanism in the distal convoluted tubule is uncertain. Phosphate reabsorption is inhibited in the early proximal tubule by volume expansion with saline, but there may be a more distal reabsorptive site (at some point beyond the last portion of the proximal tubule accessible by micropuncture) that is sensitive to PTH and unaffected by saline volume expansion.
The effects of acid-base balance on proximal tubular transport of phosphate are complex.115 Acute metabolic acidosis does not affect renal tubular reabsorption of phosphate, but chronic metabolic acidosis results in decreased proximal tubular transport, an effect possibly mediated by glucocorticoids. Respiratory acidosis decreases and respiratory alkalosis increases proximal tubular reabsorption of phosphate. Volume expansion increases urinary phosphate excretion and causes natriuresis because phosphate is cotransported with sodium in the proximal tubule.
Recently, additional factors that impact renal handling of inorganic phosphorus have been identified. Phosphatonins are circulating substances that increase renal loss of phosphorus. More than one has been identified. To date, they include fibroblast growth factor-23 (FGF-23), secreted frizzled-related protein (sFRP-4), fibroblast growth factor-7 (FGF-7), and matrix extracellular phosphoglycoprotein (MEPE).149 These substances decrease sodium phosphate transporters in proximal convoluted tubules, whereas FGF-23 and sFRP-4 are believed to also decrease formation of 1–25 dihydroxycholecalciferol. FGF-23 is increased in people with chronic renal failure, but the exact cause is unknown. Intestinal phosphatonins (factors released from the intestines due to increased intraluminal phosphate concentration) have also been suggested to exist. Although believed to be important in some pathologic conditions, their impact in normal phosphorus homeostasis is currently uncertain.
Hypophosphatemia
Clinical effects of hypophosphatemia
Hypophosphatemia can occur even when total body phosphorus is normal. Nonetheless, severe hypophosphatemia can have many detrimental effects. The most severe cellular damage seems to occur when there is concurrent phosphate depletion.99 Hypophosphatemia decreases erythrocyte concentrations of ATP, which increases erythrocyte fragility, leading to hemolysis. Hemolysis usually is not observed until serum phosphorus concentration decreases to 1.0 mg/dL or less. Hypophosphatemia also reduces erythrocyte 2,3-DPG concentrations, which impairs oxygen delivery to tissues. Leukocytes in hypophosphatemic patients have impaired chemotaxis, phagocytosis, and bacterial killing.38 This altered function may promote sepsis in hypophosphatemic patients receiving total parenteral nutrition. Platelet-associated abnormalities include shortened survival time, impaired clot retraction, megakaryocytosis in the bone marrow, and thrombocytopenia. Hemolytic anemia, thrombocytopenia, and impaired clot retraction occurred in starved dogs that were made hypophosphatemic by infusion of amino acids, ostensibly because of depletion of cellular ATP stores.178 Clinically, hemolysis has been reported in hypophosphatemic dogs and cats with diabetic ketoacidosis, hepatic lipidosis, and other disorders.2,80,173 Hemolysis was reported in four other hypophosphatemic diabetic cats, but cause and effect were obscured by the possibility of Heinz body anemia.23
Neuromuscular effects of hypophosphatemia include weakness and pain associated with rhabdomyolysis, as well as anorexia, vomiting, and nausea secondary to intestinal ileus.88,89 Decreased phosphate may impair central nervous system glucose use and ATP production, leading to metabolic encephalopathy, which has a wide range of manifestations in people (e.g., coma, seizure, confusion, irritability).99,177 Reversible impairment of cardiac contractility occurs in dogs with experimentally induced hypophosphatemia and in people with naturally occurring hypophosphatemia.65,66,180 Hypophosphatemia also causes proximal tubular bicarbonate wasting, reduction in titratable acidity, and impaired renal ammoniagenesis. However, serious acid-base disturbances do not arise in phosphate-deprived dogs.151 Phosphate deficiency produces bone demineralization via effects of PTH and calcitriol, and release of carbonate from bone may prevent serious metabolic acidosis. Hypomagnesemia frequently is found in hypophosphatemic people, but the reasons for this association are not clear.32
Causes of hypophosphatemia
Hypophosphatemia may be caused by translocation of phosphate from extracellular to intracellular fluid (maldistribution), increased loss (decreased renal reabsorption of phosphate), or decreased intake (decreased intestinal absorption of phosphate).99,136 Clinical conditions associated with hypophosphatemia are presented in Box 7-1. In confusing cases, one can measure fractional urinary phosphorus excretion to help determine if renal losses are responsible.
Box 7-1 Causes of Hypophosphatemia
Translocation related to administration of a carbohydrate load (e.g., 5% dextrose infusion) is a common cause of hypophosphatemia in hospitalized people.15,79 Insulin facilitates entry of glucose and phosphate into cells, where glucose is phosphorylated to glycolytic intermediates. Interestingly, infusion of a higher concentration (e.g., 10% dextrose) for a shorter time seems to be less detrimental than infusing 4% glucose continuously.99 Malnourished patients receiving total parenteral nutrition are particularly susceptible to hypophosphatemia because of the accelerated rate of tissue repair as phosphate is incorporated into new cells and phosphate use during glycolysis.88,136 Hypophosphatemia as part of the “refeeding syndrome” (i.e., severe electrolyte changes in malnourished patients that are being fed parenterally or enterally) was more likely in patients that were more severely emaciated, had lower initial serum phosphate concentrations, and experienced more aggressive initial infusion of parenteral nutrition.107 Respiratory alkalosis likewise causes translocation because it stimulates glycolysis by activating phosphofructokinase.88 This effect has been demonstrated in experimental dogs but was marked only when hyperventilation was combined with glucose administration.19 Increased intracellular pH may be more important than increased extracellular pH for causing hypophosphatemia in respiratory alkalosis, which could explain why severe hypophosphatemia may occur in people with severe respiratory failure who are mechanically ventilated.99
Diabetic patients are especially at risk for hypophosphatemia. They often have total body phosphate deficits because of a loss of muscle mass, urinary phosphate losses, and impaired tissue use of phosphate related to insulin deficiency. Most diabetic cats in one study had mild hypophosphatemia at presentation, whereas 20 of 48 ketotic cats in another study were hypophosphatemic.23,146 Another study found only 7 of 104 diabetic cats to be hypophosphatemic. However, stratification of the cats into ketoacidotic and nonketoacidotic groups revealed that 5 of 38 ketoacidotic cats were hypophosphatemic and only 2 of 66 nonketotic cats were hypophosphatemic.39 Interestingly, serum phosphorus concentrations are often normal to increased at presentation in diabetic people, perhaps because of metabolic acidosis by organic acids (e.g., β-hydroxybutyrate), insulin deficiency, osmotic effects of hyperglycemia, or renal insufficiency.86,119
Administration of large doses of insulin makes hypophosphatemia even more likely in diabetic ketoacidotic patients. Severe hypophosphatemia has been reported in dogs and cats treated for diabetic ketoacidosis.2,23,173 Hypophosphatemia developed or worsened after insulin administration, and clinical signs (e.g., hemolysis, seizures) thought related to hypophosphatemia developed in 11 animals. Interestingly, four of these cats developed hemolytic anemia despite intravenous supplementation of potassium phosphate, and it is not clear whether the anemia was caused by inadequate phosphate supplementation or Heinz body formation.23
Although it is not documented in dogs and cats, hypophosphatemia may occur in people with certain rapidly growing tumors. Ostensibly, the rapidly dividing cells use phosphorus, removing it from the blood.99
Increased urinary loss of phosphorus often produces moderate hypophosphatemia in primary hyperparathyroidism, but clinical signs are caused by hypercalcemia.* If 2.5 mg/dL is considered the lower limit of normal, serum phosphorus concentration was decreased in approximately one third of reported cases associated with parathyroid adenoma, but in six of six cases associated with parathyroid hyperplasia.44 Hypophosphatemia is seen inconsistently in cats with primary hyperparathyroidism.43,82 The fractional excretion of phosphorus (FEPi) was increased in a few affected dogs.172 The normal FEPi was found to be 7.5% ± 4.6% in 10 normal dogs but 10% to 23% in a dog with primary hyperparathyroidism.29
Fanconi syndrome in basenjis is associated with decreased renal fractional reabsorption of phosphate, but serum phosphorus concentrations are normal.18 The renal tubular transport abnormality may be caused by metabolic or membrane defects affecting sodium transport, and the observed phosphaturia may be secondary to natriuresis.109 Loop diuretics (e.g., furosemide) and distally acting diuretics (e.g., thiazides) have little effect on renal phosphate excretion, but proximally acting diuretics (e.g., carbonic anhydrase inhibitors) may increase renal excretion of phosphate secondary to their effects on proximal tubular sodium reabsorption. In one study, acetazolamide (10 mg/kg intravenously three times daily) did not cause hypophosphatemia when administered to dogs over a 7-day period.137 Eclampsia in the bitch may be associated with hypophosphatemia and hypocalcemia.8,10 Presumably, increased PTH secretion in response to hypocalcemia leads to decreased renal reabsorption of phosphate.
Not reported in veterinary medicine, cranial trauma is associated with renal losses of phosphorus and hypophosphatemia.132 Acquired diabetes insipidus has been suggested as a possible reason.
Hypophosphatemia caused by dietary deficiency is unlikely in animals eating commercial diets with adequate protein content. A low-protein, low-phosphorus diet designed to dissolve struvite calculi (Prescription Diet S/D, Hill’s Pet Nutrition, Inc., Topeka, Kansas.) did not cause significant hypophosphatemia when fed to dogs over a 6-month period.1 Urinary phosphorus excretion decreased and calcium excretion increased in this study. Although vomiting and malabsorptive diseases potentially can cause phosphate loss, these disorders rarely cause hypophosphatemia in dogs or cats.31 Canine malabsorptive intestinal disorders often are characterized by hypocalcemia related to hypoalbuminemia, but serum phosphorus concentrations typically are normal.20,55
People have become hypophosphatemic after administration of magnesium- and aluminum-containing antacids.101 Whether phosphate depletion occurs depends on the patient’s phosphorus intake, dosage of the phosphate binding agent, duration of administration, and the preexisting phosphate balance of the patient. Vitamin D deficiency may cause hypophosphatemia because hypocalcemia increases PTH secretion, which increases renal phosphate excretion. Decreased intestinal phosphate absorption presumably also plays a role in this setting.
It has been stated that 38% of hyperadrenocortical dogs have hypophosphatemia, but actual serum phosphorus concentrations were not reported.125 In one study, an identifiable cause of hypophosphatemia could not be found in the majority of dogs with this serum biochemical abnormality.31 Hypophosphatemia, hypercalcemia, hyperglycemia, azotemia, hypokalemia, and acidosis have been reported in a dog and cat with hypothermia caused by exposure to low environmental temperature.138 The mechanisms responsible for these electrolyte and acid-base disturbances are uncertain, but translocation seems likely.
Renal transplantation has been associated with hypophosphatemia in people and cats.123 Ostensibly, the cause is decreased renal tubular reabsorption of phosphorus, which is currently believed to be due to phosphatonins. There was no obvious consequence of the hypophosphatemia in a report of 32 affected cats. Hemolysis occurred, but the incidence was not statistically different from normophosphatemic renal transplant cats. Changing the diet from a phosphorus-restricted diet to a maintenance diet plus supplementing sodium phosphate orally corrected the problem.
Disorders of renal tubular phosphate transport associated with hypophosphatemia in humans include X-linked hypophosphatemia, autosomal dominant hypophosphatemic rickets, oncogenic hypophosphatemic osteomalacia, and hereditary hypophosphatemic rickets with hypercalciuria.168 Naturally occurring mutations in the npt2 gene encoding the type IIa sodium-phosphate cotransporter have not been identified in these disorders, but rather mutations have been found in other phosphate-regulating genes. X-linked hypophosphatemia is caused by a mutation in the PHEX gene (i.e., phosphate-regulating gene with homology to endopeptidases on the X chromosome), which is expressed in bone, whereas autosomal dominant hypophosphatemic rickets is caused by a mutation in the FGF-23 gene, a member of the fibroblast growth factor family. Oncogenic hypophosphatemic osteomalacia occurs as a result of secretion of a humoral phosphaturic factor secreted by neoplastic cells. Hereditary hypophosphatemic rickets with hypercalciuria is similar to X-linked hypophosphatemia and autosomal dominant hypophosphatemic rickets except that it is associated with appropriately increased serum concentrations of calcitriol, whereas the other hereditary disorders are not. Renal tubular disorders of phosphate transport have not been conclusively identified in dogs and cats, but hypophosphatemia, increased urinary FEPi, low serum 25-hydroxycholecalciferol concentration, osteopenia, and pathologic fractures were reported in a young cat believed to have abnormal renal tubular phosphate transport and defective hepatic 25-hydroxylation of vitamin D.74
Cats with decreased serum concentrations of cobalamin and folate have been found to be at increased risk for hypophosphatemia.135 Most of the hypophosphatemic cats had gastrointestinal tract disease or pancreatitis. Therefore, it is not clear whether the changes in cobalamin and folate were epiphenomenon or cause-and-effect. However, as these patients frequently require nutritional supplementation (either enteral or parenteral), the need to avoid the refeeding syndrome was noted.
Postsurgical hypophosphatemia is a well-established problem in people. It is more common and can be particularly severe after hepatic surgery, especially major resection and transplantation.41 Although the mechanism was once suggested to be uptake of phosphorus by the regenerating liver, excessive renal losses are now considered to be responsible.116 “Phosphate diabetes” is the term used for increased urinary phosphate excretion due to decreased phosphate reabsorption, especially in hypophosphatemic patients.96 The occurrence of hypophosphatemia after hepatic surgery in particular is not surprising because acute hepatic damage due to any number of causes also is associated with hypophosphatemia. In fact, hypophosphatemia in people with hepatic damage has been suggested to reflect healing and regeneration of the liver.144 Hypophosphatemia has been seen in dogs experimentally intoxicated with xylitol,176 but it is uncertain whether hypophosphatemia is due to hepatic damage or to other metabolic effects of xylitol.
Hypophosphatemia previously has been associated with sepsis and gram-negative infections in people to the extent that some have suggested that it should be a diagnostic tool.171 Hypophosphatemia is believed to occur in these people due to redistribution of phosphorus into body cells. Recently, severe hypophosphatemia (<1 mg/dL) has been reported to be a major risk factor for mortality in these patients.154 Decreased ventricular stroke work with subsequent decreased arterial pressure is believed to be caused in part by the hypophosphatemia.
Intravenous administration of saccharated ferric oxide to treat iron deficiency has caused hypophosphatemia in people.155 Similar changes have not been reported in dogs or cats.
Treatment of hypophosphatemia
Phosphate supplementation seems appropriate for asymptomatic patients deemed at risk for developing symptomatic hypophosphatemia (e.g., diabetic ketoacidotic cat with serum phosphorus concentration of 1.6 mg/dL) and patients with clinical signs believed to result from hypophosphatemia. The clinician should keep in mind that oversupplementation (especially but not exclusively parenteral) can cause morbidity (e.g., hypocalcemia, soft tissue calcification, renal failure). Interestingly, treatment of asymptomatic hypophosphatemia in diabetic people is controversial and recommended only when hypophosphatemia is severe (<2.0 mg/dL).85 However, clinical experience in veterinary medicine suggests that anticipatory phosphorus supplementation is reasonable in some ketoacidotic cats.
If oral supplementation is suitable (which is uncommon in patients with severe hypophosphatemia), it may be safer and therefore preferable to parenteral supplementation.5,112 Oral phosphate administration is unacceptable in vomiting patients and perhaps in patients with diarrhea. If the enteral route is chosen, feeding skim or low-fat milk or a buffered laxative usually is effective. Patients symptomatic because of hypophosphatemia generally need parenteral replacement therapy. Administering phosphate intravenously is potentially dangerous because it may cause hypocalcemia, tetany, soft tissue mineralization, renal failure, or hyperphosphatemia.88 Therefore, phosphorus administration typically has consisted of injecting small amounts slowly over hours to days and monitoring the patient repeatedly (e.g., 0.01 to 0.06 mmol/kg/hr in dogs and cats with measurement of serum phosphorus concentration every 6 to 8 hours).80,173 Although such caution is wise, it is noteworthy that more aggressive phosphorus administration has been used in people (i.e., 0.16 to 0.64 mmol/kg over 4 to 12 hours in patients receiving total parenteral nutrition).32 Other groups have used similarly large doses over even shorter times (e.g., 0.4 to 0.8 mmol/kg depending on the degree of hypophosphatemia over 30 minutes in patients with cardiac disease), also without problems.180 Sodium phosphate and potassium phosphate are commonly used, but administration of glucose phosphate also has been reported.180 Selection of the particular form of phosphorus to administer is based on the patient’s serum electrolyte concentrations.
Currently, it seems safest to administer phosphate by constant-rate infusion at rates that have been used successfully in dogs and cats and to monitor the serum phosphorus concentration every 6 to 8 hours. Theoretically, adding phosphorus to fluids containing calcium may cause precipitation of calcium phosphate, but this appears to depend on relative concentrations of calcium and phosphorus. Phosphorus usually is administered after diluting it in physiologic saline solution. The volume of distribution for administered phosphate varies tremendously among hypophosphatemic people, and redistribution of phosphate can occur rapidly. Therefore, the dose necessary for repletion and the patient’s response to therapy cannot be predicted. In two studies of hypophosphatemic cats, total amounts of phosphorus infused intravenously ranged from 0.138 to 1.26 mmol/kg, indicating a wide range of total body phosphate deficits.2,80
A conservative approach is to assume that intravenously administered phosphate remains in the ECF compartment (actually much of it enters the intracellular fluid). Development of hyperphosphatemia is unlikely with this approach. Prophylactic parenteral phosphate therapy (such as may be used for patients with diabetic ketoacidosis) may be reasonably estimated by giving one fourth to one half of the supplemented potassium as potassium phosphate and the rest as potassium chloride. However, decreased urinary phosphate excretion that develops during hypophosphatemia may persist during treatment and predispose to hyperphosphatemia. The products available for oral and parenteral use are summarized in Tables 7-1 through 7-3.

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


