Chapter 11 Respiratory Acid-Base Disorders
Control of alveolar ventilation
The drive to breathe originates within respiratory centers of the medulla (i.e., ventral respiratory column, VRC), which consist of a network responsible for respiratory rhythm generation and respiratory pattern formation. Although supramedullary brain structures, such as the pons, cerebellum and forebrain, modulate breathing, they are not required to breathe. Respiratory rhythm generation and pattern formation are altered by homeostatic control mechanisms that permit flexibility in the respiratory control system (i.e., plasticity). For example, the medullary respiratory centers are modified by sensory (i.e., chemoreceptors and mechanoreceptors) and modulatory projections (i.e., serotonergic neurons), as well as many other processes that affect breathing (i.e., cortical inputs, cardiovascular alterations, etc.). These inputs collectively establish the spatiotemporal efferent signals that project to respiratory pump muscles such as the diaphragm and upper airway muscles involved in airway resistance regulation. Altogether, these pathways result in muscular contraction, which subsequently drives alveolar ventilation50 (Figure 11-1).
Chemoreceptors and chemoreflexes
Chemoreceptors provide sensory inputs to the respiratory control system concerning changes in CO2 or O2. As a result, alveolar ventilation is altered via negative feedback mechanisms to minimize variations in normal gas levels.6,19,50,55 The primary stimuli for changes in alveolar ventilation are hypoxemia (Pao2 <60 mm Hg) and carbon dioxide-induced changes in intracellular and extracellular pH, although the magnitude of these changes is dependent on the specific chemoreflex. For example, arterial CO2 levels are tightly regulated under normal ventilatory control; increases in arterial CO2 levels (Paco2) of 1 to 3 mm Hg will double alveolar ventilation. In contrast, arterial O2 pressures (Pao2) can change up to 20 mm Hg with little alteration in ventilation.50
In adult mammals, chemoreceptors are located in both the peripheral and central nervous systems. The most significant oxygen-sensitive chemoreceptors are primarily found within the carotid body at the bifurcation of the internal and external carotid arteries. Although carotid body chemoreceptors sense changes in CO2 and pH as well, these chemoreceptors dominate the hypoxic ventilatory response and are responsible for approximately 90% of the increase in minute ventilation seen with hypoxemia.14,18 Oxygen-sensitive receptors are also found in the aortic bodies within the aortic arch. However, they appear to play a minimal role during normal ventilation.50 In addition to the periphery, O2-sensitive chemoreceptors are also present in the CNS, but their contribution to ventilatory control remains unclear.73
In contrast to O2-sensitive receptors, the primary CO2/pH-sensitive receptors are located in the CNS throughout the brainstem (i.e., retrotrapezoid nucleus [RTN], serotonergic and GABAergic neurons in the raphe nuclei, noradrenergic neurons in the locus coeruleus, nucleus of the solitary tract [NTS], pre-Bötzinger complex, and cerebellar fastigial nucleus).50 Although both peripheral and central chemoreceptors contribute to CO2-induced ventilatory responses, the central receptors appear to be quantitatively more significant for producing changes in ventilation mediated by CO2; resection of the peripherally-located carotid bodies only leads to a small increase in resting Paco2 levels (2 to 4 mm Hg) secondary to a decrease in alveolar ventilation.41,64 The CO2-sensitive chemoreceptors are extremely responsive as small changes in CO2/pH affect breathing dramatically. For example, in resting awake humans, a 1-mm increase in Pco2 increases ventilation by approximately 20% to 30%.19
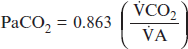 (1)
(1)where CO2 is the metabolic production of carbon dioxide and 0.863 is a constant that equates dissimilar units for CO2 and A.83 The A is the fraction of the total minute ventilation (E) that is produced at the gas exchange units of the lung. The portion of gas not reaching the gas exchange areas of the lung is termed dead space gas. The dead space to tidal volume ratio (VD/VT) refers to the portion of each tidal volume that ventilates dead space and is “wasted.” Therefore, the alveolar gas equation can be rewritten as:
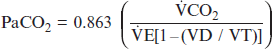 (2)
(2)As determined by equations (1) and (2), measuring the Paco2 provides direct information about the adequacy of alveolar ventilation. Many primary respiratory acid-base disorders subsequently result from alveolar hypoventilation (increased Paco2) or alveolar hyperventilation (decreased Paco2).
Mechanoreflexes
In addition to chemoafferent alterations of ventilatory control, ventilation is also influenced by sensory inputs originating from the chest wall, pulmonary and airway receptors*. Many receptors located throughout the airways are activated in response to changes in pressure, cold, irritation, and stretch. For example, laryngeal stimulation by negative pressure or cold results in reflex activity directed at maintaining upper airway patency whereas airways below the larynx contain slowly (SAR) and rapidly (RAR) adapting stretch receptors and bronchopulmonary C fibers which, upon activation, result in many reflex events including bronchoconstriction or bronchodilation, protective reflexes (i.e., cough), and alterations in respiratory timing.37
Gas diffusion and transport during respiration
Carbon dioxide
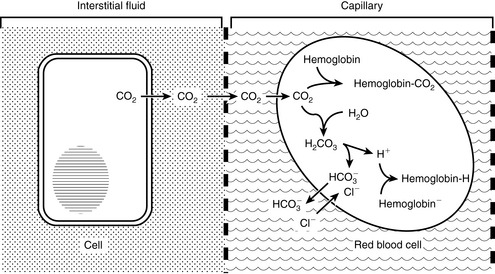
As oxygen is transported to and used by tissues, metabolic processes in the body normally produce approximately 15,000 mmol of carbon dioxide daily. The lungs are responsible for excreting a great deal more carbonic acid (H2CO3 and dissolved carbon dioxide) each day than the kidneys.63 Hence, alveolar ventilation and carbon dioxide removal have a large influence in acid-base balance. Dissolved carbon dioxide is approximately 20 to 24 times more soluble than oxygen. It is so diffusible that we can assume complete equilibration of Pco2 across membranes. As the tissues produce carbon dioxide, equilibrium is rapidly achieved between intracellular and extracellular compartments. Thus CO2 diffuses rapidly from the tissues into red blood cells. In the blood, bicarbonate is formed through the following reaction:
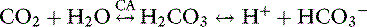
The first reaction is quite slow in plasma but fast within the erythrocyte due to the presence of carbonic anhydrase (CA), which quickly hydrates CO2 to form carbonic acid. As shown in Figure 11-2, carbonic acid spontaneously dissociates into H+ and 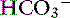 at intracellular pH. When the concentration rises,
at intracellular pH. When the concentration rises, 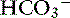 ions diffuse from the red cells into the plasma. However, the cell membrane is relatively impermeable to cations (H+), and chloride (Cl−) ions diffuse into the red cells from plasma to maintain electroneutrality (so-called “chloride shift”). In the lungs, the shift of chloride out of red cells is facilitated by the high intracellular concentration of chloride (approximately 60 mEq/L) when compared with other cells. Most of the carbon dioxide (approximately 81%) is subsequently transported to the lung as bicarbonate. A small amount is transported still dissolved in plasma (approximately 8%), and some is combined with terminal amino groups of blood proteins (approximately 11%), the most important of which is carbaminohemoglobin.78
ions diffuse from the red cells into the plasma. However, the cell membrane is relatively impermeable to cations (H+), and chloride (Cl−) ions diffuse into the red cells from plasma to maintain electroneutrality (so-called “chloride shift”). In the lungs, the shift of chloride out of red cells is facilitated by the high intracellular concentration of chloride (approximately 60 mEq/L) when compared with other cells. Most of the carbon dioxide (approximately 81%) is subsequently transported to the lung as bicarbonate. A small amount is transported still dissolved in plasma (approximately 8%), and some is combined with terminal amino groups of blood proteins (approximately 11%), the most important of which is carbaminohemoglobin.78
Oxygen
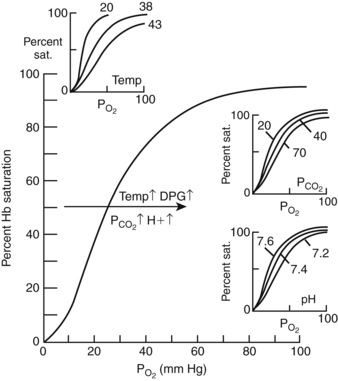
Although respiratory blood-gas disorders primarily result from alterations of CO2 levels, consideration must be also be given to alterations in O2 levels as patients with respiratory acid-base disorders may also become hypoxemic. Oxygen transport is initiated by contraction of the diaphragm with consequent movement of inspired gas down the continually branching airways until the transitional and respiratory bronchioles, alveolar ducts, and alveoli are reached. Within this respiratory zone, alveolar ventilation and gas exchange occur as oxygen moves down its concentration gradient and into the red blood cells. The partial pressure of oxygen in the red blood cells approximates that of alveolar gas within the first third of the lung capillaries, primarily due to the lung’s considerable diffusion capabilities. Oxygen is then carried in the blood to meet the oxygen demand of the tissues in two forms: dissolved and combined with hemoglobin. Most of the delivered oxygen is bound by hemoglobin with only a small contribution from the dissolved oxygen (0.003 mL dissolved O2 per 100 mL of blood/mm Hg Po2). The maximal amount of oxygen that can be combined with hemoglobin is called the oxygen capacity. Approximately 1.36 mL of O2 can combine with 1 g of hemoglobin. Assuming 15 g of hemoglobin per 100 mL of blood, approximately 21 mL O2 per 100 mL blood is carried to the tissues. As determined by the oxygen-hemoglobin dissociation curve (Figure 11-3), at low Po2, the amount of oxygen carried by hemoglobin increases rapidly with increases in Po2. However, at a higher Po2 (>60 to 70 mm Hg), the curve flattens off and little additional hemoglobin loading occurs. Unloading of large amounts of oxygen from hemoglobin is facilitated in the tissues where oxygen pressures are much lower (10 to 60 mm Hg) and the curve is very steep. Several factors shift this curve to the right and aid in the unloading of oxygen to the tissues, including increased H+ ion and carbon dioxide concentrations (as seen in respiratory acidosis), increased temperature, and increased 2,3-diphosphogycerate (2,3-DPG), a compound that competes with oxygen for its binding site on hemoglobin.

The alveolar-arterial oxygen gradient
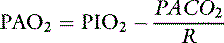
where R is the respiratory exchange ratio that accounts for the difference between CO2 production and O2 consumption at steady state, Pio2 is the inspired oxygen tension, and Paco2 is the alveolar Pco2. In normal animals, R is approximately 0.8. Because of the high solubility of CO2, Paco2 can be substituted for Paco2 in equation (4) under the assumption that Paco2 will equal Paco2.
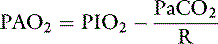
Thus the difference between PAO2 and Pao2 can be calculated as:
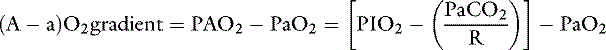
Considering R = 0. 8, and 1/0.8 = 1.25:
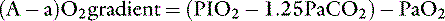
At sea level in a patient breathing room air, Pio2 is approximately 150 mm Hg. This can be substituted in equation 7:
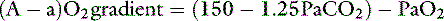
Values below 15 mm Hg are generally considered normal.16 If the (A − a) O2 ratio is widened, a component of the hypoxemia results from ventilation-perfusion mismatching. It should be remembered that Fio2 is dependent on barometric pressure and will be lower at higher altitudes. At an altitude of 500 m (approximately 1640 feet), Pio2 = 140 mm Hg, whereas at 1000 m (3280 feet), Pio2 = 130 mm Hg. Although it has long been thought that in the hypercapnic patient the alveolar-arterial oxygen difference differentiates hypoxemia caused by pure hypoventilation from hypoxemia in which other factors play a role, this idea has been seriously challenged,26,33 because the (A − a) O2 gradient may be increased in some patients with extrapulmonary disorders. Clinically, a normal gradient excludes pulmonary disease and suggests some form of central alveolar hypoventilation or an abnormality of the chest wall or inspiratory muscles.67 To increase the specificity of the test to diagnose the ventilation/perfusion mismatch, only patients with (A − a) O2 gradient values greater than 25 mm Hg should be considered abnormal.16 These patients are likely to have primary pulmonary disease, but extrapulmonary disorders cannot be completely ruled out.
Hypoxemia
Arterial blood gas analysis is not only essential for determining Paco2 levels and acid-base condition of a patient; it also provides information pertaining to a patient’s oxygenation status. There are five main reasons for hypoxemia, including low fraction of inspired oxygen, hypoventilation, diffusion impairment, ventilation-perfusion mismatching, and shunt (Box 11-1).
Box 11-1 Mechanisms of Derangement in Arterial Oxygenation
Low fraction of inspired oxygen (Fio2)
Ventilation-perfusion mismatch (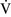 –
–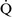 mismatch)
mismatch)
Hypoventilation
As previously discussed, the prevailing PAO2 is determined by the balance between the removal of oxygen by the blood and replenishment of oxygen by alveolar ventilation. According to equations (4) and (5) above, as alveolar ventilation decreases, PAO2 and Pao2 decrease while Paco2 and Paco2 must increase. As a result, the (A −a) O2 gradient does not change. If the (A −a) difference is widened, there may be a component of the hypoxemia attributable to primary lung disease, such as ventilation-alveolar perfusion mismatching or right-to-left shunting. In addition, the alveolar gas equation also predicts that although increases in alveolar ventilation can change PAO2 considerably, they can only moderately increase Pao2. Due to the sigmoid shape of the oxygen-hemoglobin dissociation curve, the effect of increasing alveolar ventilation on arterial oxygen saturation is minimal above a Pao2 of 55 to 60 mm Hg.27,46 Clinically, important causes of hypoventilation include CNS disease, respiratory depressant pharmacologic agents, neuromuscular diseases affecting the respiratory muscles, chest wall injury, upper airway obstruction, and severe diffuse pulmonary disease.
Ventilation-alveolar perfusion mismatch (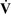 –
–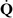 Mismatch)
Mismatch)
Despite regional differences in 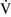 –
–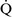 ratios throughout the mammalian lung, the heterogeneity of individual lung units is relatively limited, resulting in a
ratios throughout the mammalian lung, the heterogeneity of individual lung units is relatively limited, resulting in a 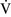 –
–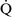 ratio of approximately 0.8.46 This ratio enables the mixed venous blood to become fully oxygenated and the CO2 to be eliminated without increases in minute ventilation.85
ratio of approximately 0.8.46 This ratio enables the mixed venous blood to become fully oxygenated and the CO2 to be eliminated without increases in minute ventilation.85
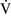 –
–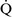 mismatch is one of the most commonly encountered causes of hypoxemia. It is present in areas of the lung where there are perturbations in ventilation or perfusion resulting in inefficient gas exchange. For example, low
mismatch is one of the most commonly encountered causes of hypoxemia. It is present in areas of the lung where there are perturbations in ventilation or perfusion resulting in inefficient gas exchange. For example, low 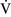 –
–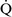 units have a low Pao2 and high alveolar Pco2, resulting in hypercapnic and hypoxemic blood. In fact, when breathing room air, the blood leaving a gas exchange unit with a
units have a low Pao2 and high alveolar Pco2, resulting in hypercapnic and hypoxemic blood. In fact, when breathing room air, the blood leaving a gas exchange unit with a 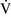 –
–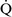 ratio of less than 0.1 is essentially unoxygenated. Low
ratio of less than 0.1 is essentially unoxygenated. Low 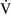 –
–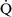 units (poorly ventilated, adequately perfused) can be found in patients with increased airway resistance (e.g., asthma, bronchitis, chronic obstructive pulmonary disease). High
units (poorly ventilated, adequately perfused) can be found in patients with increased airway resistance (e.g., asthma, bronchitis, chronic obstructive pulmonary disease). High 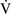 –
–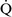 units (poorly perfused, adequately ventilated) have a high Pao2 and a low Paco2. In lung areas with
units (poorly perfused, adequately ventilated) have a high Pao2 and a low Paco2. In lung areas with 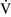 –
–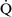 ratios greater than 1, additional increases in ventilation do not improve oxygenation.59,85 High
ratios greater than 1, additional increases in ventilation do not improve oxygenation.59,85 High 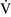 –
–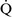 ratios are found in diseases with increased compliance (e.g., emphysema) or low output states (e.g., pulmonary embolism).
ratios are found in diseases with increased compliance (e.g., emphysema) or low output states (e.g., pulmonary embolism).
Final blood gas tensions are determined by the mixing of gas contents from different gas units. Thus 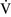 –
–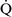 mismatch will produce hypoxemia based on the actual O2 and CO2 levels in each lung area and the amount of blood flow to each unit.59,85 The severity of
mismatch will produce hypoxemia based on the actual O2 and CO2 levels in each lung area and the amount of blood flow to each unit.59,85 The severity of  –
– mismatch can be assessed using the (A − a) O2 gradient as both abnormally low and high
mismatch can be assessed using the (A − a) O2 gradient as both abnormally low and high  –
– ratios increase the gradient. Patients with
ratios increase the gradient. Patients with 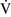 –
–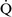 mismatch usually are hypoxemic but have normal or decreased Paco2 because chemoreceptors respond to, and minute ventilation is altered by, changes in carbon dioxide levels.58,85 Hypoxemia resulting from
mismatch usually are hypoxemic but have normal or decreased Paco2 because chemoreceptors respond to, and minute ventilation is altered by, changes in carbon dioxide levels.58,85 Hypoxemia resulting from 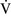 –
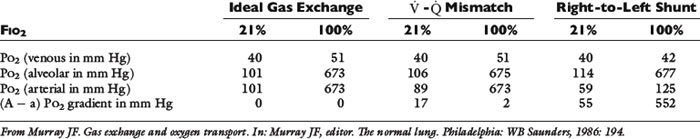
–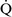 mismatch can be corrected by increasing the fraction of inspired oxygen (Fio2) by use of 100% O2.
mismatch can be corrected by increasing the fraction of inspired oxygen (Fio2) by use of 100% O2.
Right-to-left shunt
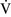 –
–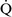 mismatch and results when mixed venous blood completely bypasses ventilated pulmonary alveoli and returns to the arterial circulation. A small amount (2% to 3%) of shunting is present in normal animals through the bronchial and thesbian circulations. In pathologic states, shunt results from perfusion of lung areas that receive no ventilation because of atelectasis or consolidation (
mismatch and results when mixed venous blood completely bypasses ventilated pulmonary alveoli and returns to the arterial circulation. A small amount (2% to 3%) of shunting is present in normal animals through the bronchial and thesbian circulations. In pathologic states, shunt results from perfusion of lung areas that receive no ventilation because of atelectasis or consolidation (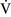 –
–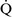 = 0) or from deoxygenated blood flow through anatomic right-to-left channels. Thus shunting is the main cause for hypoxemia in pulmonary edema, atelectasis, pneumonia, and in congenital abnormal cardiac communications between the systemic and pulmonary circulations (e.g., patent ductus arteriosus, ventricular or atrial septal defect, tetralogy of Fallot) with right-to-left blood flow bypassing the lungs.
= 0) or from deoxygenated blood flow through anatomic right-to-left channels. Thus shunting is the main cause for hypoxemia in pulmonary edema, atelectasis, pneumonia, and in congenital abnormal cardiac communications between the systemic and pulmonary circulations (e.g., patent ductus arteriosus, ventricular or atrial septal defect, tetralogy of Fallot) with right-to-left blood flow bypassing the lungs.Even small amounts of shunt result in clinically relevant hypoxemia because venous blood oxygen content is extremely low and mixed venous blood is being added directly to arterial blood without alveolar gas exchange. Similar to 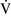 –
–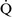 mismatch, patients with right-to-left shunting have a decreased Pao2 with a normal or decreased Paco2 and widened (A − a) O2 gradients. However, one major difference is that the Pao2 levels in animals with increased shunting fail to return to normal even with 100% O2 supplementation. In contrast, animals with
mismatch, patients with right-to-left shunting have a decreased Pao2 with a normal or decreased Paco2 and widened (A − a) O2 gradients. However, one major difference is that the Pao2 levels in animals with increased shunting fail to return to normal even with 100% O2 supplementation. In contrast, animals with 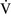 –
–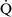 mismatch, hypoventilation, or diffusion impairment exhibit pronounced increases in Pao2 with oxygen enrichment (Table 11-1).
mismatch, hypoventilation, or diffusion impairment exhibit pronounced increases in Pao2 with oxygen enrichment (Table 11-1).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


