Fig. 1.
WHO pathological grading and molecular characterization of adult GBMs. (a) Diffuse gliomas are pathologically graded by the WHO. Grade I astrocytomas, also known as pilocytic astrocytomas, are found in children and NF1 patients, and most often remain indolent. Grade II astrocytomas or low-grade astrcytomas (LGA) tend to occur in younger adults, but do progress to higher grades. Grade III astrocytomas or anaplastic astrocytomas (AA) demonstrate increased cellularity, nuclear atypia and neoangiogenesis. Grade IV astrocytomas or glioblastoma multiforme (GBM) include features of AAs plus areas of geographical necrosis (N) surrounded by pseudopalasading cells (P) and increased aberrant neoangiogenesis (V). (b) On a clinical and molecular, but not pathological basis, GBMs can be classified as “secondary” which occur in minority of cases, or “primary” which are most common and have a worse prognosis. Secondary GBMs progress from LGA to AA and harbour mutations in IDH and aberrations in p53 and Rb cell cycle pathways, along with alterations in PDGFR signalling. Primary GBMs are characterized by amplifications and mutations of EGFR, and to a higher degree loss of PTEN. The TCGA GBM project looking at a large number of GBMs have further refined the molecular characterization to four groups, with prognostic and associated genetic alterations.
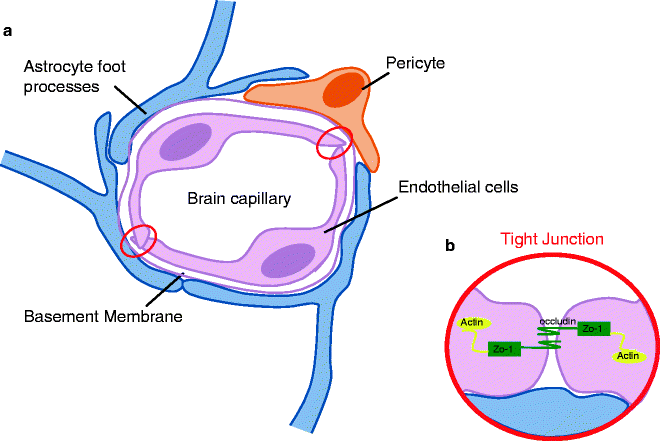
Fig. 2.
Cytostructural basis of the blood–brain barrier (BBB). (a) The integrity of BBB is reliant on the close proximity of the capillary endothelial cells and the formation of tight junction, which are more abundant in the CNS than any other system in the body. The endothelial cells are surrounded by a thick basement membrane, which in turn is surrounded by numerous astrocytic foot-end processes. The presence of pericytes in the BBB is thought to aid the interactions between the astrocytes and the endothelial cells (50). (b) Tight junctions form between two cells when their membranes are in a suitably close proximity forming a nearly impenetrable barrier. The proteins underlying the junction assembly are not fully understood due to the limitations of the molecular size; however, it is known that zonula occludens (ZO-1) is the protein which attaches to the underlying cellular actin structure. From the ZO-1 protein, many occludin molecules branch and it is these strands that are thought to assemble directly in the intercellular space to firmly bind the junction.
The metabolic demand of rapidly proliferating GBM tumour cells is reliant on an adequate blood supply that allows the continual delivery of oxygen, nutrients and growth factors. The neoangiogenic process is also involved in efficient removal of metabolic end products along with diffusion into the cerebrospinal spinal fluid (CSF), since the CNS unlike other organs does not have lymphatics. Although there are several CNS primary tumours which are highly vascularized, increased and abnormal neoangiogenesis correlates with the increasing malignancy and poor prognosis in gliomas as ultimately seen in GBMs (7, 8). The ability of tumours to induce the formation of their own blood supply to sustain their growth is a hallmark of all solid cancers (9). The growth and progression of tumours has shown to be significantly reduced in the absence of neoangiogenesis (10, 11). This has led to intense efforts aimed at understanding the underlying mechanisms driving tumour vessel formation, with the hopes of developing drugs to inhibit or destroy the vasculature of the tumour.
Highly vascularized tumours such as GBMs have increased EC proliferation and blood vessel heterogeneity (2) which could relate to their mechanisms of vessel formation. As a result of various cytokines released by glioma cells the structure of the BBB (Fig. 2) including endothelial cell tight junctions are disrupted resulting in haemorrhage and oedema (12). Although increasing EC proliferation has been shown to affect GBM vasculature, the underlying mechanisms initiating vessel formation remain poorly understood. Several mechanism(s) of neoangiogenesis have been demonstrated or proposed (Fig. 3). Initially, the tumour cells physically migrate towards blood vessels in response to signals within the tumour microenvironment and grow alongside existing vessels, a indirect mechanism of neoangiogenesis termed “vessel co-option”. By far, the most studied and understood mechanism(s) of blood vessel formation is “angiogenesis,” a process that initiates the sprouting and elongation of existing vessels into the tumour area (13). In response to oncogene activation and/or metabolic stress (nutrient deprivation and hypoxia) tumour cells secrete growth factors (GFs) such as VEGF and Angiopoietin-2 (Ang-2) (14–18). These GFs bind to their cognate receptors on nearby ECs on pre-existing blood vessels to first induce vessel destabilization followed by EC proliferation, migration and differentiation; thus forming a new blood vessel at the site of destabilization. Another mechanism of neoangiogenesis is “vasculogenesis,” a process whereby bone marrow derived progenitor cells (BMDPCs) are recruited to the area and differentiate into ECs and other vascular cell types to produce de novo blood vessels (19–21). Until recently, the process of vasculogenesis was thought to occur primarily during embryonic development but recent studies have suggested that vasculogenesis may also occur in adulthood in response to pathological stimuli such as tumour growth (19). Vasculogenesis is more prevalent in larger tumours where an increasing level of hypoxia is seen. In these tumours, increasing HIF1α signalling initiates the release of stromal derived factor 1 (SDF-1) from tumour cells, a known chemo-attractive factor which enhances the migration of bone marrow derived cells (22). Recently, there have been suggestions that tumour cells themselves can form vascular channels to transport blood independently of EC lined vessels, a phenomenon referred to as “vascular mimicry” (13, 23, 24).
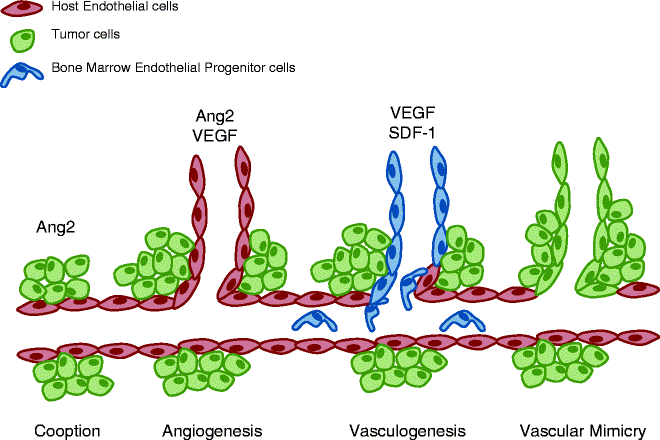
Fig. 3.
Mechanisms of neoangiogenesis: co-option, angiogenesis, vasculogenesis and vascular mimicry. Tumour progression is heavily reliant on neoangiogenesis for many metabolic aspect of growth. As such there are many mechanisms underlying vessel formation. Co-option allows tumours to invade areas where extensive networks of vessels already exist, limiting the effort the tumour needs to go through. In larger tumours, it becomes necessary to extend existing networks further and three mechanisms exist: Angiogenesis is the process of direct branching of the existing vessels, initiated by a sudden increase and angiopoietin and VEGF signalling from the tumour. Vasculogenesis is a more involved process in which endothelial precursor cells are recruited to the tumour area and in turn differentiate into the endothelial cells required to form new vessels. This process is usually observed in larger more hypoxic tumours where increasing VEGF coincides with an increase in HIF1α signalling, which in turn increases stromal derived factor (SDF-1) release, a stimulant for bone marrow derived cell migration. Vascular mimicry is the most recent hypothesis for derivation of tumour vessels. Although conflicting reports exists, it is thought that particularly aggressive tumour cells are capable of trans-differentiating into endothelial cells to form their own vessels.
2 Methods for Assessing Brain Tumour Angiogenesis
Solid tumour formation and prognosis is intimately related to its vascularization, hence understanding the mechanism(s) by which the vessels are formed and how they develop will potentially lead to define new drug targets aimed at tumour neoangiogenesis. In addition to growth and progression, tumour neoangiogenesis also is a determinant of tumour sensitivity/resistance to current non-specific therapies such as radiation (RTx) and chemotherapy (CTx), perhaps related to issues around drug delivery as well as oxygen and nutrients to the tumour cells themselves (25). In addition, the tumour initiation cells, including brain tumour initiation cells (BTICs) in GBMs, have been speculated to have a special relationship with the vasculature, residing more in the “vascular niche” (26). For these reasons, angiogenesis assays are becoming a more functional and important prognostic factor in many human cancers, demonstrating a patient’s potential to respond to treatment and the likelihood of tumour recurrence (27–30).
Due to the inaccessibility of tumours and their vasculature, many of the well-established assays of angiogenesis are set up for in vitro studies. It has therefore become essential to develop novel assays and systems for in vivo real-time studies to increase the reliability for human tumours. The aim of this section is to highlight the major angiogenesis assays used in the past decade and to further demonstrate the novel methods being developed for future use.
3 In Vitro Assays
A significant portion of our understanding of angiogenesis and vascular biology to date has come from in vitro assays which focus particularly on the proliferation, differentiation and migration of ECs in culture. Although these in vitro methods allow the user a greater degree of control over the variables within the experimental situation, the pseudo-environment created by the assay limits the extrapolation of the results to in vivo situations.
3.1 Tubule Formation Assay
Kubota et al. (31) developed the tubule formation assay more than 20 years ago. It is based on the ability of ECs to rapidly attach and align themselves into capillary-like tubules when exposed to the basement membrane and other extra-cellular matrix (ECM) components. The tubules formed have basic vessel characteristics including a lumen, adherens and tight junctions and luminal–abluminal cell polarity (Fig. 4).
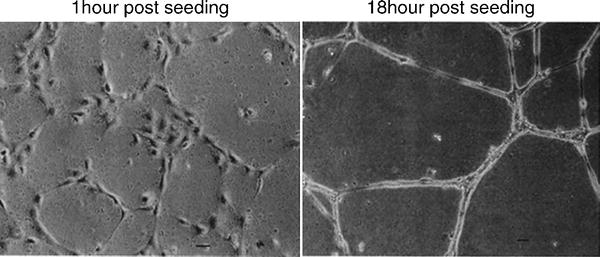
Fig. 4.
Endothelial cell tubule assay. Following initial seeding of 4,800 cells/cm2 on to matrigel substrates, HUVECs (human umbilical vascular endothelial cells) develop visible tubules within 4 h, which go onto become more defined. Adapted from (31).
Tubule Formation Protocol
2.
Dilute Matrigel 1:100 in ice-cold phosphate-buffered saline (PBS).
3.
Add 50 μl of diluted Matrigel solution to each well of a pre-chilled sterile 96-well plate.
4.
Incubate 1 h at 37°C to allow the Matrigel solution to form a gel.
5.
Harvest endothelial cells and resuspend in desired culture medium, supplemented with desired angiogenesis factors at 1–2 × 105 cells/ml.
6.
Add 150 μl of cell suspension (1.5–3 × 104 cells) per well directly onto the solidified Matrigel.
7.
Incubate the assay plate at 37°C for 4–18 h.
8.
Examine the endothelial tubes using a light microscope at high magnification. It is possible to visually estimate the extent of tube formation by inspecting the overall tube length and branch points (see Note 2). Staining with Haematoxylin and Eosin post-fixation can be done if needed.
Adapted from (33).
3.2 Adapting the Tubule Formation Assay to Cancer Biology
The basic assay described above can be adapted in order to examine the angiogenic potential of both the tumour microenvironment and the tumour cells themselves. First, in order to assess the angiogenic potential of the tumour microenvironment, components of the microenvironment can be included in the tubule formation assay through either the direct addition of tumour stromal fluid to the Matrigel substrate or through the use of tumour cell conditioned media obtained from tumour cell cultures. By examining the effect the addition has on endothelial tubule formation when compared to the standard protocol, it will elucidate how permissive to angiogenesis the tumour cells are. Those tumours which have an increased propensity to vascularize will have more heavily branched tubules in the assay. For example, conditioned media obtained from cancer stem cells (CSCs) have been shown to significantly increase the length of tubules formed during an 18-h culture, compared to ECs receiving non-conditioned media (34, 35).
Second, the tubule assay has been used to directly assess the potential of cells derived from the tumour to differentiate into tubules, so demonstrating the presence of endothelial precursors. GBM-derived BTICs are thought to contain an endothelial precursor subpopulation based on their expression of vascular markers such as CD144. When plated onto Matrigel, they align and differentiate into tubules of a similar phenotype to those seen in endothelial cell cultures (36). The presence of endothelial progenitors within tumours suggests that one of the vascular mechanisms arises from the tumour being able to generate its own vessels directly.
3.3 Cell Migration Assay
The migration assay is based on the motility of ECs and their ability to migrate through a porous membrane in a double layer culture system. The assay can not only look at different cells and their migratory characteristics but also look at cellular components that may stimulate or inhibit migration during a short period in culture (37). It is essential to know the cell characteristics because if left long enough all cells will migrate but not due to the stimulus. The Boyden Chamber technique is most widely used, but laboratories have tried adapting the procedure to include beads and other base substrates in the lower compartment. The technique in recent years has been improved with the commercial availability of kits containing prefabricated assay plates which house a specifically designed porous insert (Cell Biolabs) (33, 38) (Fig. 5).
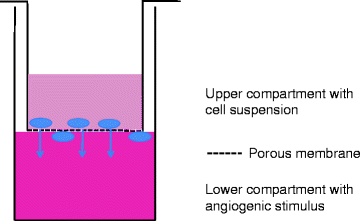
Fig. 5.
Cell migration assay setup. To test the chemo-attractant properties of a substance, a cell migration assay can be set up with the cells grown on a porous membrane and the test substance underneath. The assay is quantified as the number of cells which have moved through the membrane after 48 h of culture. Different matrix can be applied to the top chamber, such as myelin, to evaluate migration through relevant organ-specific stroma such as those found in the brain.
Cell Migration Protocol
1.
Dissolve Collagen IV gel matrix in culture media.
2.
Collagen is plated over the porous membrane inserts in the culture vessels.
3.
Plates are incubated at 37°C for 1 h.
4.
Angio-attractants or repellents are dissolved in culture media to the correct concentration.
5.
The angio-attractants or -repellents are plated in the lower culture chamber of the chosen culture vessel.
6.
The collagen coated porous insert is then placed on top.
7.
Cells of interest are harvested and resuspended at 1–2 × 105 cells/ml in media.
8.
Add 150 μl of cell suspension (1.5–3 × 104 cells) per well directly on to the porous insert.
9.
The assay plate is then incubated at 37°C for up to 24 h depending on the culture system in use.
10.
Non-migrated cells that remain on the top surface of the membrane can be scraped and lysed for genetic or protein analysis.
11.
Migrated cells are fixed and stained with Haematoxylin and independently counted in ten high-power fields.
3.4 Cell Migration Expected Results
Upon completion of the assay, a comparison can be made between the cells remaining in the upper chamber or lower chamber through genetic analysis or by direct cell counting. Increasing the concentration of angiogenic stimuli in the lower chamber should increase the number of migratory endothelial cells that reach the lower chamber and so angiogenic factors can be equated by looking at cell number.
3.5 Adapting the Cell Migration Assay to Cancer Biology
Similar to the tubule formation assay, components from the tumour microenvironment can be introduced to the lower compartment to assess their ability to stimulate migration either in the form of extracts from the primary tumour or through media conditioned by tumour cells in culture.
It has been shown that cells derived from tumours such as GBM are able to increase the degree of migration of ECs in culture (34, 35). As before, the assay can be used to demonstrate the degree of migratory signals expressed by tumours to which endothelial cells respond, indicating the potential of tumour tissue to recruit and generate new vessels.
4 In Vivo Experimental Systems
Although studying vascular biology through in vitro cell culture methods has provided a good basic understanding of their structure, minimal functional and mechanistic information can be gained until they are studied in their natural environment. Due to limitations in obtaining primary human tumour tissue, it is essential to have experimental systems and animal models for use in studying these diseases. This also gives the opportunity to study the vasculature as a dynamic process and not a static event, elucidating further the mechanisms involved in the progression of tumour development and vessel formation. Currently, there are different types of animal models in use for the study of tumour biology and each has its own advantages and limitations.
4.1 Xenografts
Xenotransplantation is the transplantation of living cells, tissues or organs from one species to another. Such cells, tissues or organs are called xenografts. In the absence of large amounts of primary human tumour samples, well-established models which utilize minimal biopsy tissue or cell suspensions have become the gold standard in assessing tumour development in situ (35, 40–42). It should be noted that these xenograft models require immunocompromised models, which is an important component of tumour–stromal interaction important in regulating neoangiogenesis.
Xenografts can be implanted orthotopically (in the same anatomical location as the source of tissue) or heterotopically (at a different location), in order to look at the effects on the tumour in both environments. In most cases, heterotopic sites are located subcutaneously in the hind flank of the mouse where it is possible to propagate primary tumour tissue samples to expand the limited amount of tumour tissue available. Injections into either orthotopic (intracranial) or subcutaneous sites in studies of GBMs include established human GBM cell lines or freshly disassociated GBM explants, the latter reportedly retaining many of the molecular and growth characteristics of the human GBMs (43, 44). In addition, these GBM lines can be transfected with a luciferase reporter to facilitate repeated bioluminescence imaging (BLI) to monitor tumour growth and response to therapeutic intervention (Fig. 6).
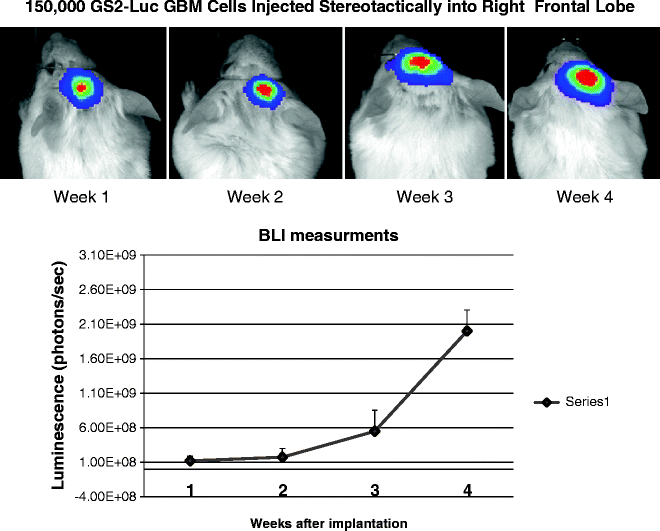
Fig. 6.
Bioluminesence imaging (BLI) of intracranial GBM xenografts. Through the integration of a Luciferase constructs to the cells of interest, it is possible to visualize the tumour volume and area following injection of the substrate Luciferin to the mouse. Through a time course study, the level of luminescence is seen to increase with time following implantation, and through these kinetics studies it is possible to determine the equivalent tumour volume and allowing evaluation of various therapeutic interventions.
Xenograft Subcutaneous Protocol
1.
Immunocompromised mice 8–10 weeks old are anaesthetized with isofluorane at 4% for induction followed by 1.5–2% throughout the procedure with 0.5–1 μl O2 per min. The mouse should be placed on a sterile towel and TearGel applied to the eyes to prevent corneal dehydration and abrasion.
For Tissue:
1.




Once anaesthetized, the mouse is laid on its side.
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
