and William A. Weiss2
(1)
The W.A. Weiss Lab, University of California, San Francisco, San Francisco, CA, USA
(2)
Departments of Neurology, Pediatrics, and Neurological Surgery, University of California, San Francisco, San Francisco, CA, USA
Abstract
Medulloblastoma, the largest group of embryonal brain tumors, is highly aggressive, with a dismal prognosis for high-risk patients. This disease has historically been classified into five variants based on histopathology. Recent genetic, epigenetic, and transcriptional analyses have categorized tumors into 4–6 subgroups, which are often inconsistent with histopathology subclasses. Mouse models of medulloblastoma are key to improving our understanding of disease progression and developing targeted therapies for patients. In this chapter, we describe different genetically engineered mouse models, and how they fit in the molecular and histopathological subclasses of human medulloblastoma. We discuss the strengths and weaknesses with which these mouse models mirror human disease, highlighting how insights from these models have informed initiation and progression of disease. Lastly, we review targeted therapeutic agent testing in some of these models and discuss future steps and goals in medulloblastoma modeling and research.
Key words
MedulloblastomaGenetically engineered mouse modelsMolecular subgroupsTargeted therapiesPreclinical testing1 Introduction
Medulloblastoma, a highly aggressive embryonal neuroepithelial tumor of the cerebellum (WHO grade IV tumor), is the most common malignant brain tumor in children (1, 2). Multimodal treatment—surgical resection, radiation, and chemotherapy—has improved overall survival rates significantly over the last decade. Five-year survival rates are now as high as 70–80% in standard-risk patients (3–5). However, this survival benefit is achieved at the cost of long-term treatment-induced morbidities, principally neurocognitive and endocrinological (6–9). Moreover, there is continued risk of secondary tumors, relapse, and metastasis (10). The prognoses for high-risk patients, especially patients less than 3 years of age with leptomeningeal dissemination at presentation, remain dismal at only 25–40% 5-year event-free survival (11, 12).
1.1 Clinical Presentation
Medulloblastoma accounts for about 20% of all pediatric central nervous system tumors (13), with an incidence of about 0.6 per 100,000 children in patients of 0–19 years (14–16). The peak age is about 7 years and is 1.6 times more common in males than females (14, 17). The incidence of medulloblastoma decreases with age: about 70% of cases occur in children younger than 10 years. Incidence progressively decreases below 0.07 per 100,000 persons above 35 years of age (16). Pediatric medulloblastoma commonly arises as a tumor of the midline cerebellar vermis; while in older patients, tumors tend to occur within the cerebellar hemispheres (18, 19).
Current risk stratifications are based broadly on age, degree of surgical resection, and disease spread. About 30% of cases display leptomeningeal dissemination at presentation, the strongest predictor of poor prognosis (20, 21). Further, the delicate proximity of medulloblastoma to the brainstem affects the extent of surgical resection which, together with the degree of tumor spread, is highly predictive of patient outcome (20, 22). Completely resected tumors in patients older than 3 years with no leptomeningeal spread at time of diagnosis are classified as standard risk, while all others are considered high risk (1, 9). Leptomeningeal spread is graded M0–M4, where M0 represents no evidence of metastasis, M1 shows some microscopic cells in the CSF, M2–M4 represents increasing metastasis from the cerebellar space to the spine and outside of the cerebrospinal axis, respectively (2).
Medulloblastoma is a heterogeneous disease comprising of four molecular and five histopathological variants. Improved stratification is required to better predict prognoses and to direct rational treatment strategies that maximize cure and minimize adverse effects. Identification of risk groups also enables development of targeted therapy based on distinct molecular subtypes and should improve clinical trial design and enrollment.
In this chapter, we will first describe the classification of different medulloblastoma subtypes based on their histopathology and molecular differences, and the genetically engineered mouse models that have been developed. We then discuss how well these mouse models mirror human disease, and how these models have contribute to the developmental therapeutics.
2 Classification of Medulloblastoma
2.1 Histopathological Features
Medulloblastoma is classified as a World Health Organization (WHO) grade 4 tumor, defined as a neoplasm that arises from the posterior fossa (23). Medulloblastoma tumor cells are aggressive in nature: they can invade into the cortex and white matter and spread via the cerebrospinal fluid in the leptomeninges and to the spine. The majority of medulloblastoma tumor cells have an undifferentiated stem/progenitor cell appearance. The 2007 WHO classification separates medulloblastoma into five variants based on their histopathological features: (i) classic, (ii) desmoplastic/nodular (D/N), (iii) medulloblastoma with extensive nodularity (MBEN), (iv) large-cell medulloblastoma, and (v) anaplastic medulloblastoma (24) (Fig. 1). D/N and MBEN tumors have better prognoses than classic tumors, while large cell and anaplastic tumors show the worst prognoses with higher frequency of metastasis. Historically, atypical teratoid and rhabdoid tumors (AT/RT) were grouped together with medulloblastomas, as these tumors were also small round blue cell tumors by histology that often develop in the cerebellum. However, AT/RTs are now felt to represent a distinct subgroup of small round blue cell tumors that lack expression of the hSNF5/INI1 tumor suppressor gene and are distinctly aggressive (25).
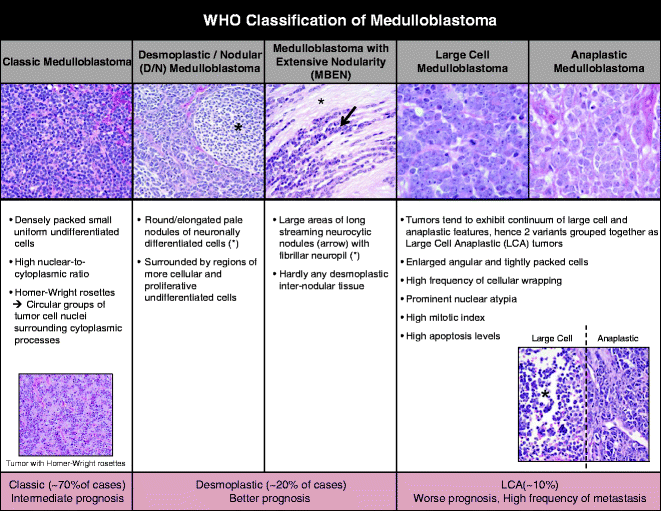
Fig. 1.
World Health Organization (WHO) classification of medulloblastoma. Medulloblastoma is divided into five variants based on their histopathological features (from left): (i) classic, (ii) desmoplastic/nodular (D/N), (iii) medulloblastoma with extensive nodularity (MBEN), (iv) large-cell medulloblastoma, and (v) anaplastic medulloblastoma. Images adapted from Eberhart, Brain Pathology (2011) and Eliison, Acta Neuropathol (2010).
Classic subtype tumors (70% of cases) are made of densely packed small uniform undifferentiated cells with high nuclear-to-cytoplasmic ratio. Homer–Wright rosettes, circular groups of tumor cell nuclei surrounding cytoplasmic processes, observed in a subset of classic tumors. Large cell and anaplastic features tend to form a continuum and are thus often grouped together. Large cell anaplastic (LCA) medulloblastoma (10%) have enlarged angular and tightly packed cells, prominent nuclear atypia, a high mitotic index, high levels of apoptosis and cellular wrapping (24, 26). Desmoplastic tumors, comprised of closely related D/N and MBEN subgroups, make up about 20% of all cases. D/N tumors appear as round or elongated pale nodules of neuronally differentiated cells surrounded by regions of more cellular and proliferative undifferentiated cells. MBEN tumors are made up of many large areas of long streaming neurocytic nodules with fibrillar neuropil and barely any desmoplastic internodular tissue (27).
2.2 Chromosomal and Molecular Biomarkers
Several biomarkers showing prognostic significance have been identified for specific clusters of tumors with some also validated in clinical trials.
1.
SHH pathway activation
The Sonic Hedgehog (SHH) signaling pathway is required for normal cerebellar development (24). During cerebellar development, a progenitor population, cerebellar granule neuronal precursors (CGNPs), proliferates and migrates across the top of the cerebellar anlage to form the external granule layer (EGL) (Fig. 2a). Large Purkinje neurons lying deep in the EGL secrete SHH, a ligand that binds its receptor Patched (PTCH1) on CGNPs. SHH binding to PTCH in turn releases the seven-transmembrane protein Smoothened (SMO) from PTCH1 inhibition. SMO subsequently activates a cytoplasmic intermediate protein complex, which results in release of the GLI family of transcription factors from their suppressor complexes that include suppressor of fused (SUFU) (13, 24). GLI transcription factors initiate downstream target gene expression and activity, stimulating the CGNPs to proliferate in the EGL (Fig. 2b). The CGNPs then terminally differentiate and migrate inwards, leaving behind a cell-sparse molecular layer, and granule-neuron dense internal granular layer (IGL). The EGL will thus cease to exist by postnatal day 15 (P15) in mice and by 1 year in humans (28–30).
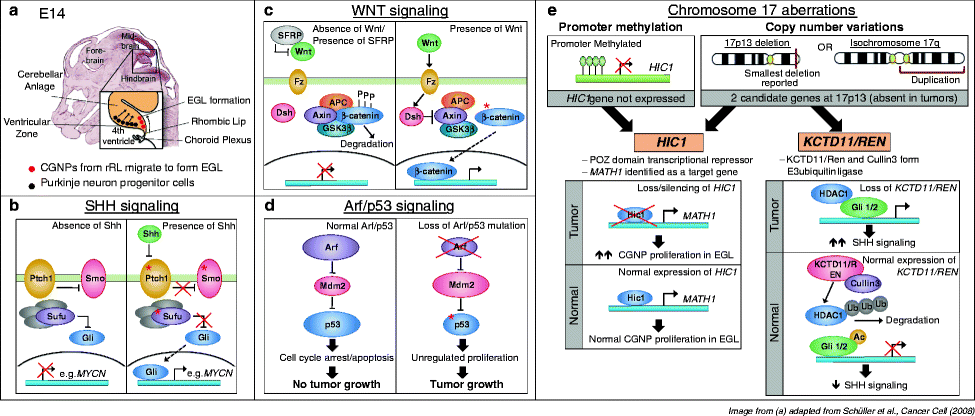
Fig. 2.
Signaling pathways and chromosomal aberrations in medulloblastomagenesis. (a) Development of the murine cerebellar anlage at E14. Cerebellar granule neuron precursors (CGNPs, in red) arise from the rhombic lip at the posterior edge of the cerebellar anlage and begin to migrate over the top surface of the anlage to form the external granule layer (EGL). Purkinje neuron precursors (in black) arise from the ventricular zone adjacent to the fourth ventricle, and migrate radially upwards through the anlage towards the EGL. Purkinje neurons are the source of Sonic Hedgehog (SHH) ligand, which activates SHH signaling in CGNPs. Image adapted from Schüller et al., Cancer Cell (2008). (b) SHH signaling pathway. In the absence of SHH ligand, Patched (PTCH1) inhibits Smoothened (SMO) to prevent downstream signaling. Suppressor of Fused (SUFU) is a negative regulator of GLI transcription factor, binding to and preventing the translocation of GLI to the nucleus, where it activates downstream target genes. SHH binding to PTCH releases inhibition of SMO, enabling GLI-driven transcription. Activation of SHH signaling in CGNPs stimulates their proliferation in the EGL. Red asterisks indicate members of the pathway that have been found mutated in human medulloblastomas. (c) WNT signaling pathway. In the absence of WNT ligand, or when SFRP family of WNT inhibitors sequesters WNT from its receptor Frizzled (Fz), the AXIN/GSK-3β/APC complex phosphorylates β-catenin, resulting in its degradation. Hence, downstream target genes are not activated. WNT ligand binds to Fz, leading to the activation of Disheveled (Dsh). Activated Dsh inhibits the AXIN/GSK-3β/APC complex, allowing β-catenin to enter the nucleus to activate downstream signaling. Activating (stabilizing) mutations of β-catenin are often found in medulloblastoma patients with activated WNT signaling [as indicated by red asterisk] (d) p53 pathway. In response to stress such as DNA damage, the transcription factor p53 can activate target genes that induce cell cycle arrest or apoptosis of the damaged cell. In the nucleolus, the Arf tumor suppressor binds to and inhibits Mdm2, which is a negative regulator of p53. Mdm2 binds to p53 to directly inhibit p53 transcriptional activity, and also has E3 ubiquitin ligase activity, targeting p53 for export out of the nucleolus into the cytoplasm for degradation. However, in tumor cells with Arf loss, Arf loss releases inhibition on Mdm2, which negatively regulates p53. This results in unchecked proliferation that leads to genetic instability in tumor cells. Mutations in p53 (asterisk) are also commonly found to result in failure to induce cell cycle arrest or apoptosis in these tumor cells. (e) Chromosome 17 aberrations. Copy number variations in chromosome 17 are present in a significant number of medulloblastoma, typically due to the formation of isochromosome 17q from the duplication of the 17q arm. This results in the gain of 17q, with a corresponding loss of 17p. The smallest deletion reported is at 17p13. Two candidate genes have been identified: Hypermethylated in Cancer 1 (HIC1) and KCTD11/REN. In addition to being deleted, HIC1 is also frequently hypermethylated at its promoter and its expression is hence silenced in tumors. HIC1 is a transcriptional repressor and MATH1 has been identified as a target gene. Hence, absence of HIC1 permits MATH1 expression and induces proliferation of the CGNPs. KCTD11/REN cooperates with Cullin3 target HDAC1 for degradation. HDAC1 deacetylates Gli1 to permit SHH signaling.
SHH pathway was first implicated in medulloblastoma when germline inactivating mutations of PTCH1 were found to cause Gorlin’s Syndrome (nevoid basal cell carcinoma) (31). These patients also showed increased incidences of rhabdomyosarcoma and medulloblastoma, suggesting that inappropriate regulation of SHH signaling in cerebellar cells contributed to medulloblastoma formation (32–34).
In about 8% of sporadic medulloblastoma cases, SHH signaling is activated due to mutational inactivation of PTCH1 (34–37). Subsequently, mutations in downstream SHH pathway components SMO and SUFU were also identified in sporadic medulloblastoma (38). The majority of SHH-dysregulated medulloblastomas present as desmoplastic or classic histopathology. Altogether, these tumors contribute to 25–30% of all cases, with desmoplastic tumors in particular associated with better prognoses (24, 39). Recently, large-scale molecular profiling has recently identified high-level amplification of additional SHH downstream effector transcription factors in a subset of medulloblastomas. This includes GLI1 and GLI2, miR17-92 complex, and YAP1, further cementing the importance of SHH signaling in medulloblastoma tumorigenesis (40–43).
2.
WNT pathway activation
The canonical WNT pathway is activated upon the binding of WNT ligands to Frizzled receptors, activating Disheveled. Activated Disheveled inhibits an AXIN/GSK-3β/APC protein complex that, in its basal state, promotes degradation of β-catenin. WNT-driven inhibition of this complex therefore stabilizes β-catenin, enabling its transport from the cytoplasm into the nucleus to activate downstream signaling (13) (Fig. 2c).
The WNT signaling pathway was first implicated in medulloblastoma from observations that a subset of patients with germline mutations in tumor suppressor APC (Turcot syndrome) developed medulloblastomas (44, 45). Subsequently, 5–10% of patients with sporadic medulloblastoma were also shown to harbor activating point mutations in the β-catenin gene CTNNB1, resulting in a degradation-resistant form of β-catenin that aberrantly activated downstream WNT signaling (46, 47). Other abnormalities found in some WNT tumors include promoter methylation (and hence gene silencing) of the secreted frizzled-related protein 1 (SFRP) family of WNT inhibitors and monosomy 6 (14, 17). SFRP1, SFRP2, and SFRP3 inhibit WNT signaling by binding to WNT ligand and sequestering WNT from its receptor Frizzled, hence epigenetic silencing of SFRP genes releases the inhibition of WNT signaling (48). Monosomy 6 is a cytogenetic marker that identifies almost all WNT tumors and tumors with monosomy 6 and β-catenin mutations associated with favorable prognosis (17, 24, 49). Comparisons between a β-catenin mouse model and human data identified a common deletion in syntenic region at human 6q25.3 encoding TULP4, which may be the candidate gene commonly deleted in the WNT tumors with monosomy 6 that is important for tumorigenesis (50). TULP4 is related to the tubby gene family and is associated with neuronal cell apoptosis. Altogether, the WNT-associated tumors account for about 18–25% of all cases and are usually of classic histology (47, 51). Patients with tumors displaying nuclear β-catenin positivity have significantly better survival (92.3%) compared to patients with tumors that are immuno-negative (65.3%), suggesting that activation of WNT, like activation of SHH (which collectively denote a minority of medulloblastoma tumors), correlates with a relatively favorable outcome (47, 51). If the association of SHH and WNT with outcome holds up, such patients may benefit from milder therapies that will reduce debilitating treatment-associated side effects.
3.
MYC/MYCN overexpression and amplification
Amplification of MYC genes (c-MYC and MYCN) correlate strongly with poor prognosis and are often found in tumors with large cell anaplastic histopathology (52). Amplification of c-MYC has been reported in 5–15% of all cases, with a small subset reported to have high-level amplification of c-MYC of >4 copies (52–54). Similarly, MYCN amplification has been found in about 10% of cases (52–54).
MYCN is a downstream target of the SHH pathway and MYCN expression levels are high in SHH-driven tumors (55–57). MYCN is the principle MYC gene that drives proliferation during cerebellar development, with little MYCN detectable after development is complete (57). MYCN levels are elevated in the majority of medulloblastoma tumors, irrespective of subtype, and overexpressing Nmyc in premalignant CGNP cells that harbor partial PTCH1 loss result in robust tumor formation (58). Further, overexpression of a MYCN transgene to the hindbrain of mice result in formation of medulloblastoma with classic and LCA histology. Over 90% of tumors that arose were demonstrated to be SHH-independent, except one mouse that harbored PTCH1 loss (59). This suggests that MYCN overexpression is sufficient to produce medulloblastoma that is both SHH dependent and independent.
4.
ARF/TP53 pathway
Li-Fraumeni patients with germline mutations in TP53 have increased risk of cancers including medulloblastoma (60). TP53 is found on chromosome 17p and was initially the prime candidate involved in chromosome 17 aberrations. However, TP53 loss or mutation contributes only up to 10–15% of all medulloblastoma (61, 62). Other members of the ARF/TP53 tumor suppressor pathway have also been implicated in medulloblastoma. Loss of ARF due to homozygous deletion or promoter hypermethylation (with wild-type TP53) has been reported in about 10% of tumors (61). An analysis of a small subset of human tumors found that TP53 mutations, methylation, and deletions of ARF are common in more aggressive LCA tumors, demonstrating that ARF/TP53 is important in promoting tumor malignancy (61).
5.
Chromosome 17 aberrations
The only frequent chromosome aberration found in medulloblastoma is isochromosome 17q, present in 30–50% of cases (4, 17). Isochromosome 17q is formed when two copies of 17q arms and two copies of centromeric 17p are fused together to form an especially long chromosome with two centromeres in place of a normal chromosome 17. This results in one copy of 17p and three copies of 17q, leading to the proposition that genes encoding for oncogenes may be present on 17q while genes encoding tumor suppressors are present on 17p. 17p losses or 17q gains have been reported as well, with loss of 17p present in up to 30–40% of cases (63–65). Altogether, copy number variations in chromosome 17, including 17q gains, isochromosome i17q, and 17p deletions, have been associated with poor prognosis (52).
The smallest region of deletion reported at 17p is at 17p13.3. Two candidate tumor suppressors have been identified at 17p13 that likely play a role in medulloblastoma tumorigenesis. These are namely Hypermethylated in Cancer 1 (HIC1) and RENKCTD11 (66–69) (Fig. 2e). HIC1 maps exactly at 17p13.3, the region of deletion most commonly found in medulloblastoma. HIC1 is a POZ domain transcriptional repressor that is frequently hypermethylated at its promoter and thus silenced in medulloblastoma tumors (68, 70, 71). RENKCTD11, working with Cullin3, forms an E3 ubiquitin ligase complex that degrades HDAC1, which is responsible for deacetylating GLI1 and GLI2 to permit SHH-downstream signaling (72). Thus, loss of RENKCTD11 during 17p deletion can result in increased deacetylated GLI1/2 to enhance SHH signaling in SHH-driven tumors.
6.
Other epigenetic mechanisms
Hypermethylation of promoter regions of other tumor suppressor genes have been identified. RASSF1A (ras association domain family protein 1, isoform A) is a tumor suppressor gene whose promoter is hypermethylated in a number of adult cancers leading to epigenetic gene silencing (73, 74). RASSF1A is involved in cell cycle control, microtubule stabilization, cell adhesion, motility, and apoptosis (75). In medulloblastomas, promoter-associated CpG island of RASSF1A was found to be extensively hypermethylated in both alleles, resulting in epigenetic silencing of RASSF1A (76). Biallelic promoter hypermethylation of tumor samples were of all histological variants and of both adult and pediatric patients, while non-tumor samples demonstrated no methylation evidence.
SPINT2 (serine protease inhibitor kunitz-type 2) is another tumor suppressor gene that acts as an inhibitor of HGF/MET signaling. SPINT2 expression was found to be silenced via promoter methylation in 34% of primary tumor samples examined (77). In addition, some tumors harbored hemizygous deletions in the SPINT2 locus and gains in regions of HGF and MET loci. Altogether, SPINT2 expression was downregulated in 73% of tumors, while MET expression was upregulated in 45% of tumors. Re-expression of SPINT2 in medulloblastoma cell lines resulted in reduced cell proliferation and motility, and improved survival in vivo in a xenograft system, supporting the potential role of SPINT2 in medulloblastoma tumorigenesis (77).
Other genes reported to be genetically and epigenetically altered include Kruppel-like Factor 4 (KLF4) and Dickkopf-1 (DKK1). KLF4 expression is absent in 40% of tumor samples; CpG methylation of KLF4 promoter occurs in about 16% of tumors, while recurrent homozygous deletions were found in rare cases (78). Accordingly, introducing demethylating agent 5-azacytidine (5-azaC) to medulloblastoma cell lines results in the re-expression of KLF4. DKK1 is a WNT antagonist whose expression is downregulated in primary samples compared to normal cerebellum (79). Treatment of medulloblastoma cells with histone deacetylase (HDAC) inhibitor results in DKK1 upregulation, suggesting that histone acetylation may downregulate DKK1 expression, reducing its tumor suppressor functions in medulloblastoma.
Caspase-8 is involved in mediating TRAIL receptor-induced apoptosis, and it was reported that caspase-8 (CASP8) promoter region is hypermethylated in 55% of primitive neuroectodermal tumors/medulloblastomas (73). However, while CASP8 expression was reduced in half of tumor samples, expression of related members in the pathway like TRAIL receptor DR4, the adapter protein FADD and caspase-3 expression was unchanged in tumor samples compared to normal brain samples. This result suggests that downregulation of caspase-8 is particularly critical in permitting medulloblastoma tumor cell growth.
Investigations also suggest a link between histone lysine methylation and medulloblastoma tumorigenesis. In one study, SNP analysis revealed aberrant genetic events that target the methylation of repressive chromatin mark on histone H3, lysine 9 (H3K9) (80). Homozygous deletions in histone lysine methyltransferase EHMT1 and SMYD4 and hemizygous or homozygous deletions of H3K9-interacting Polycomb genes L3MBTL3, L3MBTL2, and SCML2 also occur. On the other hand, amplification and gains of histone lysine demethylases JMJD2C, JMJD2B and histone lysine acetyltransferase MYST3 that regulate H3K9 methylation were found in a subset of tumors. Thus, the authors suggest that these gene aberrations may lead to H3K9 hypomethylation, contributing to the transformation in medulloblastoma. Recently, in another sequencing effort, inactivating mutations of the histone-lysine N-methyltransferase genes MLL2 or MLL3 were also identified in a small subset (16%) of patients, suggesting role of these putative tumor suppressor genes in medulloblastoma tumorigenesis (81).
7.
Other pathways implicated in medulloblastomagenesis
Notch signaling is activated in a subset of medulloblastoma, via overexpression or amplification of ligands NOTCH1 and NOTCH2, as well as receptors HES1 and HES5 (82, 83), or via silencing of miR199b-5p, a negative regulator of NOTCH (84). PI3K pathway has also been implicated in medulloblastoma tumorigenesis. Mutations in the catalytic subunit PIK3CA are found in about 5% of cases (85). Deletions or mutations in the opposing phosphatase PTEN (a negative regulator of PI3K) occurs in 30–35% of cases (86).
Developmental genes OTX2 and TRKC have been associated with medulloblastoma as well (87). OTX2, a transcription factor expressed in the posterior cerebellum in the EGL and IGL and important in normal cerebellar development, is commonly amplified particularly in non-SHH, non-WNT tumor cases and those of anaplastic histology (88–90). TRKC, a receptor for neurotrophic factor neurotrophin-3 that is widely expressed by granule cells in the cerebellum during development, is expressed in a subset of tumors and correlated with favorable prognosis, as TRKC activation promotes apoptosis and hence inhibits tumor growth (87, 91, 92). Cell cycle factor CDK6 is overexpressed in a subset and is correlated significantly with poor prognosis (93).
2.3 Molecular Profiling
Several analyses of medulloblastoma tumor samples have attempted to subclassify this disease based on transcriptomal profiles (17, 94–96). These unbiased analyses from human tumor samples reveal some parallels that can be drawn between these subclassification and the molecular lesions discussed above.
Two subclasses of tumors, with dysregulated WNT or SHH signaling, have been identified consistently (17, 94–96) (Fig. 3). The clustering of WNT and SHH tumors away from other medulloblastomas suggests that these tumors arise and develop differently from other subtypes. WNT subtype of tumors demonstrates overexpression of WNT signaling pathway members, and these tumors typically harbor monosomy 6 and activating mutations in β-catenin gene CTNNB1. WNT tumors tend to show classic pathology and patients are associated with favorable prognosis. Demographic analyses show that patients are usually older children more than 3 years (with median of about 9–10 years)(17, 94). High MYC levels and upregulated NOTCH and PDGF signaling have also been associated with this tumor subclass (17, 94).
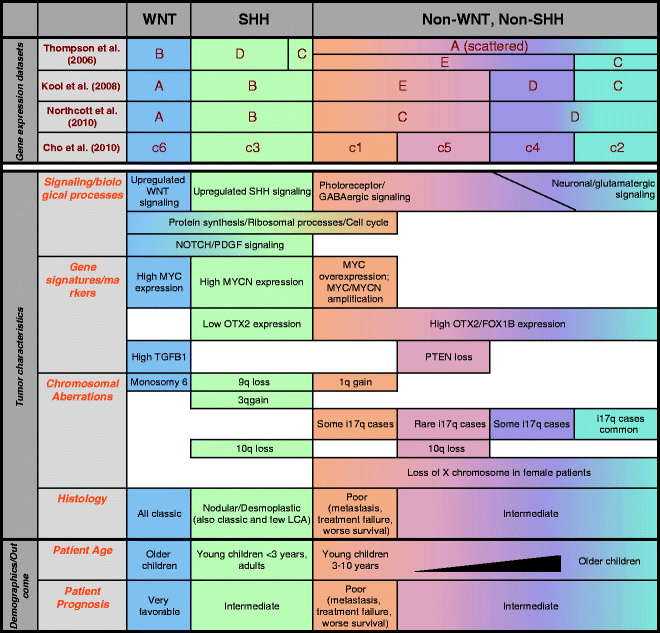
Fig. 3.
Subclassification of medulloblastomas based on transcriptional profiling. Expression profiling from four independent groups has identified WNT and SHH tumors as distinct subgroups, while subclassifying non-WNT, non-SHH tumors into 2–4 subgroups. Listed are some common features of the subgroups based on tumor characteristics and patient demographics and outcome.
SHH tumor subclass demonstrates overexpression of genes in SHH signaling pathway such as GLI1 and MYCN, and PTCH1 mutations are exclusively found in these tumors (17, 94). In addition, 9q deletions are also exclusively found in these tumors, with corresponding 9p gains (forming i9p isochromosome)(17, 94, 96). Demographic analyses reveal that patients tend to be very young children (<3 years) or adults, and these patients tend to show favorable prognoses (17). Desmoplastic tumors largely segregate in SHH subclass, although some desmoplastic tumors are found in the other subclasses (17). Similar to WNT tumors, SHH tumors have been associated with upregulated NOTCH and PDGF signaling (94). Interestingly, oncogene OTX2, which is overexpressed in a large majority of medulloblastomas, is lowly or not expressed in SHH tumors (94).
Independent groups have classified non-WNT, non-SHH tumors into 2–4 subgroups (Fig. 3). These tumors are hard to subclassify as they are more closely related. Gene expression studies have generally identified neuronal and photoreceptor markers as cluster determinants. Thompson and colleagues were the first to conduct gene expression arrays on medulloblastoma patient samples, and they classified the tumors into five subclasses A–E, of which groups B and D were the WNT and SHH subclasses, respectively (95). Of the other groups, they observed that deletion of 17p (hence i17q isochromosome) was found primarily in group C tumors, and anaplastic cases were most common in group E tumors.
A subsequent study by Kool et al. classified WNT and SHH tumors as groups A and B respectively, and divided the non-WNT, non-SHH tumors into three other clusters (94). Cluster determinants used include expression of neuronal differentiation (groups C and D) or retinal photoreceptor genes (groups D and E). Notably, metastatic disease was strongly associated with these non-WNT, non-SHH tumors, and most strongly with group E tumors. Additionally, female cases within these non-WNT, non-SHH tumors had a high incidence of loss of inactivated X chromosome. Other observations were that groups C and D weakly expressed protein synthesis and cell cycle-related genes compared to all other groups, and chromosome 17 aberrations were strongly associated with groups C or D tumors. Lastly, patients with group D or E tumors were mostly young children (<3 years), similar to SHH tumors.
A later study by Northcott et al. identified four subclasses of medulloblastomas, of which the WNT and SHH subclasses also segregated distinctly (17). The non-WNT, non-SHH tumor subgroups, groups C and D, showed expression of axon guidance and neuronal developmental genes, and also had high expression of OTX2 and FOXG1B. Group C/D tumors also harbored i17q chromosomal aberrations that were not found in WNT or SHH tumors. Notably, several of these observations mirrored findings by Kool et al. in their classification of subgroups C to E (94).
Analysis by Northcott et al. also revealed that group C tumors often amplified and/or overexpressed c-MYC, had the poorest prognosis, presented with frequent metastatic dissemination, and were represented only in young children (17). This group was also associated with genes involved in phototransduction and glutamate signaling. On the other hand, Group D tumors with no expression of MYC or MYCN, and the patients were more distributed across all age groups, particularly in adolescents. Group D tumors were associated with neuronal semaphorin signaling, G-protein coupled receptors and cyclic AMP signaling.
Lastly, the most recent study by Cho et al. analyzed the largest number of tumor samples, characterizing the mRNA and miRNA expression profiles, as well as conducted single nucleotide polymorphism (SNP) analysis for chromosomal changes (96). Six subgroups of medulloblastomas were identified, of which the WNT (c6) and SHH (c3) subgroups clustered similarly to the other published reports. Similar to previous studies, chromosome 17 aberrations were not found in the WNT or SHH subgroups.
Of the non-WNT, non-SHH subgroups, c1 and c5 were similar to each other, displaying photoreceptor/GABAergic signatures, while c2 and c4 showed neuronal/glutamatergic signatures. Comparing between c1 and c5 subclasses, upregulation/amplification of c-MYC and translational/ribosomal signatures were specific to c1. Importantly, c1 subgroup, but not c5, was associated with poor clinical outcome. Some of the signatures that distinguish c2 and c4 subclasses are as below: c2 subgroup was observed to upregulate immune-associated markers such as tumor necrosis factor α, interferon-β, and nuclear factor-κB, while c4 subgroup tumors were more enriched in MYC and MYCN. c4 subgroup was also striking in that although the tumors expressed neuronal/glutamatergic markers, they also expressed some photoreceptor markers. Recall that this result mirrors Kool’s classification of group D tumors (94).
A comparison of the broad histopathological WHO classification of classic, desmoplastic and large cell/anaplastic tumors with the molecular/transcriptional subclasses draws some parallels, but also highlights some inconsistencies in these subclassification schemes. Classic tumors are the most common subtype, but histology is not informative as these tumors span across all molecular subtypes. Desmoplastic tumors are indeed most commonly found in the SHH tumor group, but importantly, are also found in non-WNT, non-SHH tumors. LCA histopathology in tumor samples does not offer much insight, as LCA tumors can occur in all molecular subgroups; interestingly, the WNT LCA tumors have been reported to have good prognosis while a subset of non-WNT, non-SHH LCA tumors has worse prognosis. This is in line with the correlation of WNT tumors (regardless of histopathology) with good prognosis, and the correlation of high levels of MYC/MYCN (mostly group C tumors in Northcott’s analysis) with poor prognosis. Altogether, we need to employ a combination of various prognostic markers from both histopathological and molecular classifications to better stratify patients into risk groups.
3 Modeling Medulloblastoma
Research groups and pharmaceutical companies are continually developing a wide spectrum of compounds that inhibit potential targets in tumor biology. However, a big challenge is finding the most appropriate platform to screen for effective drugs against different subclasses of medulloblastoma. The majority of preclinical testing is done in tumor-derived cell lines, which are easy to use in screening assays. However, cells grown in plastic or matrix-coated plates lack cellular heterogeneity and tumor–stromal interactions and thus do not faithfully recapitulate in vivo settings.
Transplantation of human or murine cells into immunodeficient or immunocompetent mice is a popular in vivo tumor model system as highly penetrant and reproducible tumors can be induced. However, the tumors that develop do not recapitulate the sequential acquisition of changes in both tumor and tumor microenvironment changes that both moderate aspects of malignant progression. Within this context, orthotopic transplantations into immunocompetent mice are closer representations of the tumor setting over subcutaneous injections or use of immunodeficient mice, as tumors are formed in the appropriate tissue context and allow for antitumor adaptive immune responses. Also, direct implantation of fresh tumors or minimally passaged cells has been used to reduce shortcomings of the xenograft/allograft system. Regardless, results generated from these models should still be evaluated with caution.
The use of genetically engineered mouse (GEM) models provides the most physiologically relevant insight into human tumor settings, albeit not without its own limitations. GEM models initiate tumorigenesis via introduced perturbations in molecular pathways relevant to the disease. Evolving with stromal cells in the microenvironment, the tumor is more likely to mimic genetic and molecular changes and exhibit angiogenic and invasive characteristics representative of human tumors.
Historically, the first models of medulloblastoma were derived in the 1970–1980s by inoculating viral agents such as adenoviruses and polyoma viruses into rodents early postnatally during cerebellar development. One of the early successes was the use of Simian adenovirus SA7 inoculated intracerebrally in newborn hamsters that resulted in brain tumors in 22% of animals in 1–2 months, most of them resembling human medulloblastomas (97, 98). Similarly, JC human polyoma virus introduced in hamsters resulted in medulloblastomas in 3–6 months in 80–95% of animals (99, 100). The tumors were found to originate from the EGL which then migrated to the IGL (100), providing one of the first evidence in animal models of the origin of medulloblastoma. Transgenic mice engineered to express JC virus T antigen (viral early regulatory protein) also resulted in medulloblastomas in 9–13 months (101). These tumors were resembled human tumors of classic histology and presented occasional Homer–Weight rosettes, demonstrating the importance of the early viral T antigen in medulloblastoma formation. These models will not be elaborated upon in this chapter. Instead, we will focus on the more recent mouse models that have been developed using introduction or deletion of specific genes to better mimic the molecular heterogeneity of the human tumors
(Table 1).
Table 1
Mouse models of medulloblastoma
Mouse model | Tissue specific | MB tumor incidence; Latency (Lat) | Histology/tumor features | Immunoreactivity/ gene expression | Other tumors or defects observed | Molecular/chromosomal changes, other characteristics | Reference(s) |
|---|---|---|---|---|---|---|---|
SHH-driven models | |||||||
SHH pathway—germline-modified models | |||||||
Ptch1 +/− | Ubiquitous | 14–19%; Lat: 10 months (tumors seen as early as 5 weeks, peaks at 16–24 weeks) | Classic EGL expansion and compression of normal cerebellum Circumscribed, no infiltration of parenchyma/white matter | Neurofilament, synaptophysin, GFAP, reelin, trkC | Sarcomas, hydrocephalus, hindlimb polydactyly or syndactyly, larger body size | Contradicting reports in retention of remaining WT Ptch1 or LOH or mutations of remaining Ptch1 High Gli1 expression | |
Single dose 3 Gy X-ray | Ubiquitous | P1 has decreased incidence from 81% to 3% compared to P10 pups (compared to 6.7% non-irradiated Ptch1 +/− mice); Lat: 7–20 weeks P4 pups: 51% compared to 7.7% non-irradiated Ptch1 +/− mice; Lat: 38 weeks 3-month mice: no increase in tumor incidence | Similar to Ptch1 +/− non-irradiated tumors | N.D. | Sarcomas, lymphomas, BCCs | P1 pups have lower apoptosis, increased pro-survival Akt and decreased p53 with IR, compared to P10 LOH of remaining Ptch1 | |
Ptch1 +/− ; Ptch2 ±/− | Ubiquitous | 15.4–17.4% compared to 6.4% in Ptch +/− mice; Lat: 1–10 months | Similar to Ptch1 +/− tumors | GFAP, Tuj1, synaptophysin | Sarcomas, BCCs, angiectasis | Overexpression of Gli1/2, Math1, Sfrp1 LOH of remaining Ptch1 | (109) |
Ptch +/− ; p53 −/− | Ubiquitous | 95–100% compared to 14% in Ptch +/− mice; Lat: 4–16 weeks | Similar to Ptch1 +/− tumors | N.D. | Sarcomas, lymphomas, BCCs | WT Ptch1 retained in tumors Elevated Gli1 expression Random chromosomal losses | |
Conditional Ptch1 homozygous deletion | hGFAP-Cre, Math1-Cre | 100%; Lat: 1–3 months (hGFAP: 3–4 weeks; Math1: 2–3 months) | Similar to conventional Ptch1 +/− tumors, disrupted normal cerebellar architecture | Math1, NeuN, synaptophysin, GABRA6 | None | N.D. | (119) |
Conditional Ptch1 homozygous inactivation; conditional p53 deletion | Pax7-Cre | 30%; (p53 WT: Lat: 88–100 days; p53 partial/complete deletion: 32–65 days) | Similar to Ptch1+/− tumors | Tuj1, NeuN, synaptophysin | None | Invaded subarachnoid space, 50% of mice show spread to fourth ventricle | (122) |
CAGGS-CreER; R26-SmoM2 (ubiquitous) | Ubiquitous | 40%; Lat:18 weeks | Classic | NeuN, Zic1 | Rhabdomyosarcomas, BCCs, pancreatic and gastrointestinal lesions | Upregulated Shh pathway members, like Ptch1/2, Gli1 and Cyclin D1. Upregulated PDGFRα, IGF and MAPK pathways, c-Myc, N-myc, Otx2, Ezh2, Sox18, Foxm1 | (117) |
Conditional SmoM2 activation | hGFAP-, Math1-, Olig2-, Tlx3-Cre | 100%; Lat: 1–2 months (hGFAP: shorter survival) | Classic hGFAP and Math1-Cre: diffuse EGL tumors Olig2 and Tlx3-Cre: focal tumors in posterior lateral EGL lobes | GFAP, Tuj1, synaptophysin, Math1, Zic1, Pax6, Olig2 | None | N.D. | (28) |
Conditional SmoM2 activation | Gli1-CreERT2 | 40% Incidence (tamoxifen administration at P10) | Classic, Focal tumors | N.D. | None | N.D. | |
SmoA1 | NeuroD2 (ND2) | Hemizygotes—48%; Lat: 26 weeks Homozygotes—94%; Lat: 2 months | Classic, EGL hyperproliferation Homozygotes: Disruption of normal cerebellar architecture. Polygonal tumor cells, rosette formation. Necrosis, neovascularization, invasion of fourth ventricle/molecular layer, leptomeningeal spread to spinal cord and distant brain sites | Nestin, NeuN Homozygotes: loss of NeuN expression representing loss of neuronal differentiation | None | Upregulation of Gli1/2, N-myc and cyclin D1. Induction of NOTCH signaling genes such as Notch2 and HES5, and increased expression of cell cycle regulatory genes and ribosomal proteins | |
Sufu +/− ; p53 −/− | Ubiquitous | 58%; Lat: 10 months | Classic | Synaptophysin, Tuj1, GFAP | Rhabdomyosarcomas, lymphoma, thymoma | LOH of Sufu in tumors Upregulation of Math1, N-myc, Ptch2, Sfrp1, cyclin D1 Gli1/2 not upregulated | (123) |
SHH pathway—retroviral somatic gene transfer models | |||||||
SHH-MMLV, injected E13.5 | Ubiquitous | 76%; Lat: 2–3 weeks | Classic | GFAP, NeuN | None | Upregulation of Math1 | (125) |
RCAS-SHH in Ntv-a mice | Nestin tv-a | 9–34%; Lat: 3 months | Classic | Tuj1, NeuN, synaptophysin, trkC, Math1 | None | N.D. | |
SHH + IGF2 | 39% (SHH alone—15%); Lat: 12 weeks | Classic, some infiltration to brain stem | Comparable proliferation and reduced apoptosis compared to SHH alone (SHH + IGF2:Upregulated IGF pathway, phospho-AKT and N-MYC) | (131) | |||
SHH + activated AKT-Myr- Δ11-60 | 48% (SHH alone—15%) | Classic | (SHH + AKT: high levels of N-MYC) | ||||
SHH + wild-type NMYC | 47%; Lat: 47 days (SHH alone—15%; Lat: 48 days) | Classic | Increased proliferation and reduced apoptosis compared to SHH alone, high levels of cyclin D2 | (130) | |||
SHH + NMYC T50A | 78%; Lat: 22 days (SHH alone—15%; Lat: 48 days) | Classic | |||||
SHH + BCL-2 | 78%; Lat: 47 days (SHH alone—34%; Lat: 48 days) | Classic | Comparable proliferation and reduced apoptosis compared to Shh alone | (129) | |||
SHH + PTEN deletion (via PTEN-floxed) | N.D. | 70% of tumors: 100% extensive nodularity 30% of tumors: extensive nodularity with 10–30% areas with high proliferation | N.D. | (135) | |||
SHH + MYC | 23% (SHH alone—9%) | Classic, invasion into brainstem and fourth ventricle | N.D. | (132) | |||
SHH + HGF | 78% (SHH alone—39%) | Classic, some tumors with desmoplastic regions (D/N and MBEN) | Increased proliferation and reduced apoptosis compared to SHH alone | (128) | |||
Models showing upregulation of SHH pathway | |||||||
Cell cycle-associated genes | |||||||
Conditional Rb deletion in p53 floxed/null mice | mGFAP-Cre | Rb complete loss: 85–100%; Lat: 3–4 months Rb partial loss, floxed p53: 75%; Lat: 3–7 months | LCA tumors, EGL expansion from Math1+ CGNPs | Tuj1, MAP-2, GFAP, Math1 Negative for neuron-specific enolast, NeuN and synaptophysin | Lymphomas, sarcomas in p53-null background | N-myc, Ptch2 and Gli2 mutations | |
Ink4c −/− ; p53 −/− | Ubiquitous | 8%; Lat: 5 months | Classic, some LCA regions Failure of CGNP migration into IGL, hyperproliferation. Some tumors infiltrate through molecular layer into granule layer and white matter, into brain stem | GFAP, synaptophysin, nestin | Vascular tumors such as angiomas and hemangiosarcomas, pituitary ademona, B and T cell lymphoma, etc. | Overexpression of Gli1, N-myc, c-Myc, cyclin D1 | (142) |
Single dose 4 Gy γ-irradiation of Ink4c ±/− ; p53 −/− at P5-6 | Ubiquitous | 75–88% (compared to 68% in p53 −/− mice and 20% in Ink4c +/− ; p53 +/− mice) | Not mentioned, presumably similar to Ink4c −/− ; p53 −/− mice | Not mentioned, presumably similar to Ink4c −/− ; p53 −/− mice | Other tumors (not mentioned) | Not mentioned, presumably similar to Ink4c −/− ; p53 −/− mice | (147) |
Conditional p53 deletion in Ink4c −/− mice + 4 Gy γ-irradiation at P5 | Nestin-Cre | 100%; Lat: 5 months | None | ||||
Ink4c ±/− ; Ptch1 +/− | Ubiquitous | 37–46% (compared to 7% in Ptch1 +/− mice); Lat: 40 weeks | Similar to tumors from Ptch1 +/− and Ink4c −/− ;p53 −/− mice | Not mentioned, presumably similar to tumors from Ptch1 +/− and Ink4c −/− ;p53 −/− mice | N.D. | LOH of Ptch1, but no LOH of Ink4c in Ink4c heterozygotes Similar to tumors from Ptch1 +/− , Ptch1 +/− ;p53 −/− or Ink4c −/− ;p53 −/− mice: Upregulation of SHH signaling pathway members Gli1/2, Foxm1, Ptch2 High levels of Math1, cyclin D1/2, N-myc expression | |
DNA repair enzymes | |||||||
Lig4 −/− ; p53 −/− | Ubiquitous | 100%; Lat: 9 months (tumors seen as early as 3 weeks) | Classic, from EGL expansion | Synaptophysin, MAP2, Tuj1, GFAP Negative for NF | Pro B-cell lymphoma | High levels of Math1, Gli1 | (150) |
Ku80 −/− ; p53 −/− ; Rag1 −−/- | Ubiquitous | 66.7%; Lat: 4–5 months | Classic, from EGL expansion | GFAP, synaptophysin | T-cell lymphoma | N.D. | (151) |
Conditional Xrcc4 deletion in p53 −/− mice | Nestin-Cre | 100%; Lat: 25 weeks (tumors seen as early as 4 weeks) | Classic, from EGL expansion Occasional Homer-Wright rosettes 2 mice have tumors with LCA regions, 1 mouse with spinal metastasis (out of 18 mice) | GFAP, NeuN | Thymic lymphoma, solid tumors | N-myc and cyclin D2 amplification, chromosomal translocations, single-copy deletion of Ptch1 | (153) |
Conditional Brca2 deletion in p53 ±/− mice | Nestin-Cre | In p53 +/− mice: 83%; Lat: 13 weeks In p53 −/− mice: 72%; Lat: 21 weeks | Classic, from EGL expansion | N.D. | Lymphoid tumors | N-myc amplification, Ptch1 loss LOH of p53 in p53 +/− mice | (154) |
Parp-1 −/− ; p53 −/− | Ubiquitous | 49%; Lat: 16 weeks (tumors seen as early as 8 weeks) | Classic, from EGL expansion, invasive Some tumors with Homer-Wright rosettes, larger tumors invaded fourth ventricle, spread into surrounding structures | NeuN, Nestin, MAP2 Weak synaptophysin Negative for GFAP | Lymphomas, angiosarcomas | High expression of Math1 Upregulation of SHH pathway via reduction in Ptch1 and increase in Gli1 Aneuploid tumor cells Chromosomal aberrations such as fusons, chromosome/chromatid breaks, fragmentation | (152) |
Hypermethylated in Cancer 1 (Hic1) | |||||||
Ptch1 +/− ; Hic1 +/− | Ubiquitous | 40%; Lat: 25 weeks (compared to 10%; similar latency in Ptch1 +/− mice) | Classic, similar to tumors from Ptch1 +/− mice | Nestin, synaptophysin, GFAP | None | High expression of Smo, Gli1/2, Sufu Wild-type Ptch1 allele retained but not expressed Hic1 expression low or absent, Hic1 protein absent | (69) |
Overexpression of Ifng | |||||||
hGFAP-tTA; TRE-Ifng | hGFAP-tTA | E16 early induction of Ifng expression: 80%; Lat: 3–4 weeks (Ifng expression required before P16) | Classic, similar to tumors from Ptch1 +/− mice Tumors in molecular layer and EGL, also invaded into white matter and brainstem regions | Synaptophysin, Math1 | None | N.D. | (155) |
Non-SHH models | |||||||
WNT pathway models | |||||||
RCAS (c-MYC and S37A β-catenin) in Gtv-a; p53 −/− mice | GFAP tv-a | 83%; Lat: 6 weeks | LCA tumors | N.D. | None | N.D. | (156) |
Conditional β-catenin activation in p53 ±/− mice | Blbp-Cre | p53 +/− mice: 4%; Lat: 10 months p53 −/− mice: 15%; Lat: 10 months | Classic, non-EGL tumor origin Tumor formed from proliferation of Zic1 + mossy-fiber neuron precursors in dorsal brainstem/lower rhombic lip | Zic1 | None | N.D. | (50) |
MYCN overexpression | |||||||
Glt-tTA; TRE-MYCN/Luc (GLT/TML) | Glt1 | TML hemizygotes: 40%; Lat: 7 months TML homozygotes: 75%; Lat: 7 months | Classic and LCA tumors, rare metastasis to spine Non-EGL tumor origin: EGL cells in GTML mice identical to wild-type | GFAP, moderate synaptophysin, negative for NeuN and Math1 | None | No SHH pathway activation except one case with Ptch1 loss Elevated levels of Otx2 Chromosomal aberrations, particularly syntenic to human chromosome 17 | (59) |
Various GEM models have been developed using 2 main strategies: (A) germline modifications via transgenes, genetic knock-ins or knockouts, and (B) somatic cell gene transfer methods using Moloney murine leukemia virus (MMLV) or avian retrovirus RCAS/tv-a (replication-competent ASLV long terminal repeat with splice acceptor/tumor virus-A) systems. Because of the molecular and genetic complexity of medulloblastoma, it is unlikely that a single mouse model can perfectly encompass all variants of the disease. Multiple mouse models, each driven by specific but relevant genetic modifications, will together provide avenues to understand the disease.
4 Mouse Models of Medulloblastoma
4.1 SHH-Driven
Many investigations in the past have centered on the SHH signaling pathway because of its importance in normal cerebellar development and its implication in Gorlin’s Syndrome. The established link of SHH pathway with medulloblastoma has led to the several mouse models that modulate members of the pathway. These includes (a) germline-modified mouse models with partial deletion of Ptch1, activation of Smo and partial deletion of Sufu; as well as models using somatic gene transfer in which SHH and other cooperative proteins are ectopically expressed using retroviruses. Each of these will be elaborated in respective sections below.
1.
Get Clinical Tree app for offline access

Germline-modified mouse models
(a)
Deletion of Ptch1
While homozygous deletion of Ptch1 results in early embryonic lethality due to cardiac and neural tube defects, heterozygous mice are viable. Medulloblastoma tumors developed in 14–19% of heterozygous mice by 10 months, with peak occurrence at 16–25 weeks of age (102–104). Due to systemic heterozygosity for Ptch1, these mice also develop other tumors, notably sarcomas, as well as other developmental abnormalities like hydrocephalus and polydactyly. Medulloblastoma tumors arise from the external germinal layer of the posterior fossa in these mice, form well-circumscribed tumors of classic histology that compress normal cerebellar tissue but did not infiltrate the normal parenchyma or migrate along the white matter tracks. Immunoreactivity of these tumors is similar to human tumors and displays positive neurofilament, synaptophysin and glial fibrillary acidic protein (GFAP) reactivity, which are consistent with tendency of neuronal and glial differentiation of medulloblastoma tumors (104).
While decreased SHH signaling is likely to be sufficient in promoting tumor initiation, the sufficiency of heterozygous Ptch1 to promote tumor progression remains controversial. Some groups have observed the retention of wild-type Ptch1 allele in these Ptch1 +/− murine tumors (102, 104), while others report loss of heterozygosity (LOH) or mutation of the remaining Ptch1 allele (105, 106). Of note, preneoplastic proliferative lesions developed early in these mice, but most did not progress to develop full-blown tumors, suggesting that additional hits are required to achieve transformation (106).
Subsequently, several approaches were employed to increase the incidence of tumors in this model. Pazzaglia et al. demonstrated that X-ray irradiation of newborn Ptch1 +/− markedly increased incidence of tumors from 7.7% to 81% by 5–10 months (107, 108). However, tumor susceptibility is dependent on age of irradiation. In comparison to mice irradiation at P1, mice irradiated at P10 had much reduced susceptibility to tumors while mice irradiated at 3 months of age had unaltered tumor incidence (107, 108). CGNPs from P1 pups had reduced apoptosis in response to radiation via increased pro-survival AKT signaling and reduced TP53 levels compared to those from P10 pups, likely accounting for their increased resistance to irradiation (108). Further, characterization of medulloblastomas that developed from irradiated Ptch1 +/− mice revealed the LOH of wild-type Ptch1 allele, suggesting that complete loss of Ptch1 was required for increased malignancy.
Ptch2 is highly similar to Ptch1, sharing 73% amino acid similarity. Although Ptch2-deficient mice demonstrated no obvious developmental defects or tumor susceptibility, partial or complete loss of Ptch2 in a Ptch1 +/− background modestly reduced tumor latency for tumor formation and increased incidence of tumors from 6.4% to 16% compared to Ptch1 +/− mice (109). Similar to Ptch1 +/− mice, a wide range of tumors formed including sarcomas and basal cell carcinoma and there was also LOH of Ptch1 in these tumors. Altogether, these results suggest that loss of Ptch2 can cooperate with Ptch1 loss to promote medulloblastoma tumor progression.
Complete loss of p53 in Ptch1 +/− mice increased incidence of tumors (95–100%) and reduced latency to 4–16 weeks (110, 111). Combined loss of p53 and loss or haploinsufficiency for Ptch1 is proposed to (1) Deregulate the G2/M cell cycle checkpoint while also increasing genomic instability and (2) Allow cells that harbor secondary mutations to proliferate unchecked and promote tumorigenesis. Interestingly, tumors that arise from Ptch1 +/− mice did not harbor p53 mutations or deletions, suggesting that medulloblastoma formation in these mice occur independently of TP53. Tumors from Ptch1 +/− ; p53 −/− mice did demonstrate increased random chromosome loss compared to the Ptch1 +/− ; p53 +/+ mice. The tumor suppressor p19Arf indirectly activates TP53, but interestingly, combination of Arf loss with Ptch1 heterozygosity did not phenocopy Ptch1 +/− ; p53 −/− mice. Loss of Arf did not drive increased incidence of tumors in mice mutant for Ptch1, hinting at a synergistic effect between TP53 and PTCH1 in medulloblastoma tumorigenesis (110).
Loss of Ptch1 results in increased activity of downstream genes Gli1 and Cyclin D1 in CGNPs. Accordingly, the complete inactivation of Gli1 in Ptch1 +/− mice resulted in reduced incidence of tumor (3.4% of observed mice), demonstrating that GLI1 contributes significantly to SHH-driven medulloblastoma. These compound mutants upregulated GLI2, suggesting that GLI2 can partly compensate for the loss of GLI1 (112). Similar to Gli1, a complete loss of Cyclin D1 in Ptch1 +/− mice also suppressed formation of medulloblastoma (113). Lastly, Ptch1 +/− mice were crossed into an Igf2-null background, because rhabdomyosarcomas in Ptch1 +/− mice had consistently increased IGF2 levels (114). No tumors developed in the double mutant mice, demonstrating that IGF2 is essential for SHH-driven medulloblastoma (115). It was revealed that these double mutants harbored preneoplastic lesions, but these lesions failed to progress to form tumors. Further analysis showed that Igf2 transcription is not initiated until late tumor stages, demonstrating that unlike SHH signaling, IGF2 is not necessary for initial lesion formation, but is necessary for malignant progression (116).
(b)
Activation of Smo
A number of transgenic mouse models have been generated using activated SMO, either with mutation W535L (SmoM2) in human SMO or the corresponding mutation W539L in mouse Smo (SmoA1). The mutation resides in the transmembrane domain of SMO and results in SHH ligand-independent activation of downstream signaling.
The SmoM2 mouse model used a ubiquitously expressed CreER transgene to allow for tamoxifen-mediated activation of mutant SmoM2 and induction of downstream SHH signaling (117). Sporadic leakiness of Cre activity resulted in all mice developing rhabdomyosarcomas and 27% of mice developing medulloblastoma. Acute activation of SmoM2 via tamoxifen administration at post-natal day P10 increased rhabdomyosarcoma numbers per mouse, induced basal cell carcinoma-like lesions and increased medulloblastoma incidence to 40%. Medulloblastoma tumors had typical features of classic tumors, were highly proliferative and exhibited neuronal differentiation markers like ZIC1 and NeuN. These tumors also upregulated PDGFRα, components of the IGF2 and MAPK pathways, as well as markers previously implicated in medulloblastoma like c-MYC, NMYC, and OTX2.
The SmoA1 mouse model uses the NeuroD2 (ND2) promoter to drive SmoA1 expression in CGNPs (83, 118). Hemizygous mice developed medulloblastoma at a median age of 26 weeks with 48% incidence, while 94% of homozygous mice developed tumors at 2 months. Initial analyses on tumors from hemizygous mice demonstrated upregulation of SHH targets Gli1 and Nmyc, as well as concomitant induction of NOTCH signaling, suggesting that SHH and NOTCH pathways cooperate in promoting survival of tumor cells in SHH-driven tumors (83). Like other SHH-driven tumors, these tumors developed from CGNPs in the EGL. In particular, tumors from homozygous mice displayed areas of necrosis, neovascularization, and invasion into the fourth ventricle and molecular layer. Notably, these tumors often displayed leptomeningeal spread that is characteristic of high-risk patients with poor prognoses. Tumor cells were found in the leptomeninges of the brain, at distant sites from the tumor and within the gray matter of the spinal cord. Hemizygous mice also had limited frequency of leptomeningeal spread (118).
(c)




Other tissue-specific Ptch1 and Smo models
Other tissue-specific knockouts have been generated to restrict activated SHH signaling to the cerebellar neural stem cells (NSCs) or CGNPs. Utilizing hGFAP-Cre or Math1-Cre drivers to deleted Ptch1 conditionally in mice, Yang et al. observed that incidence of tumors increased dramatically to 100% by 1–3 months of age, compared to Ptch1 +/− mice (119). hGFAP-Cre mice employed the human regulatory sequences for GFAP that were reported to drive Cre in Nestin-positive radial glia cells, which generate most cerebellar cell types including CGNPs. Math1-Cre expression is restricted to unipotent CGNPs of the rostral rhombic lip (rRL), a transiently lived structure from which CGNPs are generated and migrate to form the EGL. Similar tumors developed using either driver; however, tumors initiated from NSCs with hGFAP-Cre arose at higher incidence than those initiated from CGNPs using Math1-Cre, resulting in death by 3–4 weeks compared to 2–3 months. Similar to the systemic deletion of Ptch1 and activation of Smo in mouse models described earlier, CGNPs in the EGL of Math1-Cre; Ptch1 flox/flox mice were highly proliferative at 6 weeks, with an otherwise normal cerebellar organization. This result suggests that Ptch1 loss in GCNPs induced increased proliferation and self-renewal capacity, while still permitting neuronal differentiation. By 10–11 weeks, however, full-blown tumors were present that disrupted normal cerebellar structure (119).
In a similar fashion, Schüller et al. expressed activated SmoM2 in a tissue-specific fashion using hGFAP-, Math1-, Olig2-, and Tlx3-Cre drivers (28). Olig2-Cre and Tlx3-Cre are specifically expressed in neuronal progenitors in the posterior EGL and remain expressed in IGL. Medulloblastoma developed robustly in 100% of mice using all 4 drivers, with an average survival of about 1–2 months. Like the conditional Ptch1 model, the different Cre drivers gave rise to tumors with similar classic histopathology and expression profiles. Tumors varied in extent of tumor spread, reflective of Cre expression. Medulloblastomas driven by hGFAP-Cre:SmoM2 and Math1-Cre:SmoM2 were diffuse, while Olig2-Cre and Tlx3-Cre drivers generated focal tumors restricted to the posterior lateral lobes. Like the Ptch1 model, hGFAP-Cre:SmoM2 mice had significantly shortened survival compared to the other Cre drivers. The authors also targeted SmoM2 using the Gli1 promoter. However, incidence of tumors was only at 40%, and the animals had prolonged survival, suggesting that driving tumors using promoters of genes downstream from SHH less potently promotes oncogenesis (28).
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree