Chapter 4
Principles of sedation, anticholinergic agents, and principles of premedication
Terminology
The terminology used in pharmacology and medical anaesthesia textbooks to describe drugs which act on the central nervous system undergoes regular change as understanding of the modes of action evolves. The term hypnotic is used regularly for a depressant of the central nervous system, which enables the animal to go to sleep more easily, or a drug used to intensify the depth of sleep. The animal may be aroused by stimulus. However, the term ‘hypnosis’ is also used for the ‘sleep’ component of anaesthesia (see Chapter 1). The term sedative appears to be rarely used in medical literature, but can be considered to be a drug which relieves anxiety and, as a result, tends to make it easier for the patient to rest or sleep – they are usually associated with drowsiness. Many drugs fall into both the sedative and the hypnotic categories, the differentiation usually being related to dose. They are best considered as one group, exemplified by α2-adrenoceptor agonists (α2-agonists) such as dexmedetomidine where low doses cause drowsiness and higher doses cause sleep. The term tranquillizer (or ataractic) appears now used only in the veterinary texts but previously was considered to be a drug with a predominant action in relieving anxiety without producing undue sedation and that will affect mood and behaviour.
There is considerable overlap between the action of CNS depressant drugs. Benzodiazepines are considered to be both anxiolytics and sedative/hypnotics. Drugs such as phenothiazines, now classified as antipsychotic in the medical literature, are those previously termed neuroleptics. They reduce psychomotor agitation, exerting their effects by blocking dopamine-mediated responses in the central nervous system. Overdose causes marked extrapyramidal symptoms and parkinsonian-type tremor. However, the dopamine-blocking effects of such drugs mean that they are now also considered to contribute to anaesthesia (see Chapter 1). The term ‘tranquillizer’ is still loosely used in clinical anaesthesia to cover both the anxiolytics and antipsychotics and will continue to be used in places in this and later chapters.
The multiplicity of definitions is confusing, but understanding of the major actions of the drugs is important in order to appreciate their limitations. For example, if drugs such as the benzodiazepines are used for premedication, they will not quieten a fit animal, but may make it more difficult to handle by removing its inhibitions, so that vicious animals become more likely to bite or kick. By reducing nervousness, phenothiazine derivatives may make an animal more liable to sleep, but will not make the vicious animal easier to handle, no matter what dose is given. Effective sedation depends on selection of the drug appropriate for the procedure, the species of animal, its temperament and condition, and must allow for possible side effects. Drug combinations are often more effective in controlling animals than high doses of an individual drug. Where ‘sedative’ and ‘tranquillizing’ drugs are used for premedication, low doses are usually utilized and their effects on the subsequent depth and duration of anaesthesia must be taken into account. In all cases, it is important that the animal is left undisturbed for an adequate period of time after administration of the sedative because stimulation during the onset of the drug’s action may prevent the full effect from developing. To sedate an animal that is in pain, a suitable analgesic must be used, possibly in combination with a sedative drug, because most sedative drugs themselves have little or no analgesic activity and may cause exaggerated reactions to painful stimulation.
During the past three decades, major pharmacological advances have been made in the recognition of specific drug receptor sites and of the actions resulting from their stimulation or blockade. These advances have been followed by synthesis of potent drugs, which act as agonists or as antagonists at such sites. These advances have resulted in a better understanding of the actions of existing drugs, in newer and more potent agonist drugs, and of antagonists enabling the reversal of some sedative agent effects.
Phenothiazine derivatives
This group of drugs still has a major role both in human psychiatric medicine and in veterinary anaesthesia. They are classified as antipsychotic drugs (or in older terminology ‘neuroleptics’). They are dopamine antagonists, but also block α1-adrenoceptors. The degree of activity in different pharmacological actions varies from one compound to another. All have a wide range of central and peripheral effects which have been well reviewed (Tobin & Ballard, 1979; Lees, 1979).
Being dopamine antagonists, they have calming and mood-altering (antipsychotic) effects, and also a powerful antiemetic action, particularly against opioid induced vomiting. The degree of sedation produced varies between drugs. They are metabolized by the liver, and long-term dosage can lead to liver damage. In medical practice, sedation is an unwanted side effect but, in veterinary medicine, the phenothiazine derivatives are used primarily for this purpose. In general, they are not considered to have analgesic activity, although methotrimeprazine is claimed to be a powerful analgesic in humans. Their major cardiovascular side effects are related to their ability to block α1-adrenoceptors, and thus having an antiepinephrine (antiadrenaline) effect. This results in a fall in arterial blood pressure primarily due to peripheral vasodilatation, and a decrease in packed cell volume caused by splenic dilation. Phenothazines exert an antiarrhythmic effect on the heart (Muir et al., 1975; Muir, 1981) that was originally thought to be due to a quinidine action on the cardiac membrane (Lees, 1979) but may be caused by a blocking action on the cardiac α-arrhythmic receptors (Maze et al., 1985; Dresel, 1985). The phenothiazines have a spasmolytic action on the gut although, at least in horses, gut motility is not reduced (Davies & Gerring, 1983). However, as they cause relaxation of the cardiac sphincter, in ruminants they increase the chance of regurgitation should an animal become recumbent. Phenothiazines have varying degrees of antihistamine activity and also produce a partial cholinergic block. All cause a fall in body temperature partly due to increased heat loss through dilated cutaneous vessels and partly through resetting of thermoregulatory mechanisms. In spite of all their side effects, the phenothiazines are well tolerated by the majority of normovolaemic animals.
Although promethazine is used as an antihistamine, the most commonly used phenothiazine today in the UK, North America and Australasia is acepromazine. Other derivatives such as chlorpromazine, propionylpromazine and promazine are or have been used elsewhere.
Acepromazine
Acepromazine, also known as ACP (its original trade name) or as ace, is the 2-acetyl derivative of promazine and has the chemical name 2–acetyl-10-(3-dimethylaminopropyl) phenothiazine. It is prepared as the maleate, a yellow crystalline solid. It is metabolized in the liver and both conjugated and non-conjugated metabolites are excreted in the urine. Some of these metabolites are detectable for a considerable period (McGree et al., 2013). Although available for over 50 years, it remains widely used in veterinary medicine for sedation, premedication, and a range of non-anaesthetic related indications. It continues to be the subject of a huge number of scientific publications.
Like all phenothiazine drugs, with low doses there are effects on behaviour and, as the dose is increased, sedation occurs but the dose–response curve rapidly reaches a plateau after which higher doses do not increase, but only lengthen sedation and increase side effects (Tobin & Ballard, 1979). Further increase in doses may cause excitement and extrapyramidal signs. In many animals, sedation may be achieved with intramuscular (IM) doses as low as 0.03 mg/kg, although the drug has been used safely at ten times this dose when prolonged effects were required. A calming effect on the behaviour of excitable animals can be seen at doses even below 0.03 mg/kg, making acepromazine a drug liable to abuse especially in the greyhound and the equine sporting field. The length of action is dose dependent but can be prolonged. Clinically obvious sedation lasts 4–6 hours after doses of 0.02 mg/kg. In horses, Parry et al. (1982) considered that there were detectable residual effects for 12 hours after doses of 0.1 mg/kg, and for 16–24 hours after 0.15 mg/kg IM. In general, the apparent sedation is not as great as that which can been achieved with α2-agonists, although owners of giant breeds of dog often complain that their animals are sedated for several days following acepromazine administration. Despite this, there are many situations where acepromazine is the sedative drug of choice.
In practice, the dose is chosen in relation to the length of sedation required and the purpose for which it is needed. However, the drug cannot be relied upon to give sedation in all animals; some individuals fail to become sedated and, in these, other drugs or drug combinations must be employed. Excitement reactions are rare but have been reported following intravenous (IV) or IM injection of the drug (MacKenzie & Snow, 1977; Tobin & Ballard, 1979). Other central effects of acepromazine include hypothermia and a moderate antiemetic effect, especially against opioid-induced vomiting (Valverde et al., 2004).
Clinical doses of acepromazine have little effect on respiration; sedated animals may breathe more slowly but the minute volume of respiration is unchanged. In all species, acepromazine, as all phenothiazines, causes a dose-related fall in arterial blood pressure mediated through vasodilation. The lowering of blood pressure is well tolerated in fit patients but may not be so in shocked or hypovolaemic animals. The effects of clinical dose rates of acepromazine on heart rate are generally minimal, most investigators having found a slight rise (Kerr et al., 1972; MacKenzie & Snow, 1977; Parry et al., 1982) or no change (Muir et al., 1979). However, Popovic et al. (1972) reported that, in dogs, doses of 0.1 mg/kg IM acepromazine caused bradycardia and even sinoatrial arrest. Changes in cardiac output appear to be minimal (Maze et al., 1985) even when in association with anaesthetic agents (Sinclair & Dyson, 2012).
Fainting and cardiovascular collapse have been reported to occur occasionally in all species of animal following the use of even low doses of acepromazine. In some cases, it may have been due to administration to a hypovolaemic animal but, in others, it has not been explained. Some strains of Boxer dogs (particularly those in the UK) are renowned for collapsing after a very small dose of acepromazine given by any route, and it has been suggested that this may be due to vasovagal syncope.
Acepromazine has little antihistamine activity. It has a powerful spasmolytic effect on smooth muscle including that of the gut, which may explain why it can give some visceral analgesia (Sanchez et al., 2008) and is effective in treatment of equine spasmodic colic. Acepromazine is not thought to have specific analgesic properties. Although it reduces amount of inhalation anaesthetic agents required (Heard et al., 1986; Doherty et al., 1997), it is not effective against experimentally applied nocioceptive stimuli (Sanchez et al., 2008; Bergadano et al., 2009).
Acepromazine has some very specific properties, which increase its usefulness as a premedicant agent:
•It reduces the dose required of anaesthetic agents for induction and maintenance
•It has antiarrhythmic effects and protects against epinephrine-induced fibrillation (Muir, 1981)
•When used as a premedicant in horses, it significantly reduces the incidence of death associated with anaesthesia and surgery (Johnston et al., 1995; 2002). In dogs, its use also significantly reduces anaesthetic-related deaths when compared with no premedication (Brodbelt et al., 2008a,b)
•It has been shown to be a free radical scavenger (Serteyn et al, 1999)
•Its use in anaesthetized horses reduces the ‘shunt fraction’ and increases arterial oxygenation (Marntell et al., 2005).
Relative contraindications to use of acepromazine include:
Acepromazine causes paralysis of the retractor penis muscle and protrusion of the flaccid penis from the prepuce in bulls and stallions; it is often given to facilitate examination of the penis. In horses, however, physical damage to the dangling penis may result in swelling and failure of the organ to return within the prepuce when the drug action ceases. This event, which may eventually necessitate amputation of the penis, has been reported following the use of several phenothiazine derivatives. There were several reported incidences of priapism, where there is an erection of the penis (rather than a flaccid paralysis) in stallions following administration of the neuroleptanalgesic mixture ‘Large Animal Immobilon’ which contains acepromazine (Pearson & Weaver, 1978). There are also very occasional reports of priapism occurring following the administration of acepromazine alone (van der Harst et al., 2002; Taylor & Bolt, 2011). Priapism in adult geldings has been effectively treated by the administration of 8 mg of benztropinemesylate (Wilson et al., 1991), a drug used in the treatment of Parkinson’s disease. However, this drug is not readily available. As both priapism and flaccid paralysis with subsequent physical injury are equally calamitous in valuable breeding stallions, the manufacturers specifically contraindicate use of acepromazine in these animals. The risk is very low (Driessen et al., 2011). Should priapism occur, the condition must be treated quickly and efficiently; an Esmarch’s bandage will reduce the erection and allow the penis to be returned to the sheath where it can be retained, but should this fail, or in case of a flaccid paralysis, the penis must be supported to prevent further damage.
At one time, it was thought that acepromazine might reduce the threshold at which epileptiform seizures occurred and therefore should not be used in dogs with epilepsy, or prior to myelography. With doses in the clinical range, there was no good evidence to support this theory (Tobias et al., 2006; da Costa et al., 2011; Drynan et al., 2012) and, in Europe, this contraindication has been removed from the product information.
Solutions of acepromazine for injection are available at different concentrations (country dependent). Tablets for oral use in dogs and cats and a paste for oral use in horses are also available. The injectable forms are non-irritant, painless and are effective by IV, IM or subcutaneous (SC) routes. Following IV injection, sedation is usually obvious within 5 minutes but full effects may not be apparent for 20 minutes and, when the drug is used for premedication, at least this period should be allowed to elapse before anaesthetic agents are given. Maximal effects are seen 30–45 minutes after IM and SC injection.
Very small doses of acepromazine have been used to treat behavioural problems in dogs and horses but the dose required in any individual case can only be found by trial and error. Parenteral doses of acepromazine for sedative and premedicant purposes in most domestic animals are in the range of 0.025–0.1 mg/kg. In horses, oral availability is good (Hashem & Keller, 1993) and the recommended dose is 0.15 mg/kg; this contrasts with dogs where there is marketing authorization for oral dosage up to 3 mg/kg, although whether this dose is required or is desirable is doubtful. Recommended doses for specific purposes will be discussed in the chapters relating to the individual species of animal.
Propionylpromazine
Propionylpromazine, 10-(3-dimethyl aminopropyl)-2-propionylphenothiazine, has been used widely in some countries for sedation and premedication of both small and large animal patients. Its actions, the sedation it produces and its side effects are very similar to those of acepromazine. In horses, it is used in doses of 0.15–0.25 mg/kg and, in dogs, the dose ranges from 0.2 to 0.3 mg/kg. It has also been widely used in combination with methadone.
Fluphenazine
Fluphenazine is a very long acting (several weeks) agent that has a licence for use in humans to treat certain psychiatric diseases. It has a great affinity to dopamine D2 receptors, thus acting as a dopamine antagonist. It has been used to produce long-term sedation for horses stabled through injury, but also ‘abused’ as a ‘calmer’ for horses in competition, although this is strictly against competition rules. There are no published trials relating to its pharmacokinetics or clinical use in animal species, but several detailing side effects in the horse. These can be dramatic, and include sweating, ataxia, and extrapyramidal signs, the latter being reported to continue for some days after dosing. The toxicity is well reviewed by Brashier (2006). Although this drug has no direct use in anaesthesia, if the animal has been pretreated there may be additive effects with drugs used for anaesthesia.
Other phenothiazine agents
Chlorpromazine was used extensively in veterinary practice but has largely been replaced. Its actions and side effects are similar to those of acepromazine, but it is less potent (doses of up to 1 mg/kg were used in all species of animal), has a longer duration of action and produces less sedation. Promazine has actions similar to those of chlorpromazine but is claimed to give better sedation with fewer side effects. For premedication, it was administered at doses of up to 1 mg/kg. Methotrimeprazine is a typical phenothiazine but it is also a potent analgesic, having a potency about 0.7 times that of morphine. In veterinary practice in the UK, it was combined with etorphine as the neuroleptanalgesic mixture ‘Small Animal Immobilon’. Promethazine is used in veterinary medicine primarily for its potent antihistamine activity, although recently there has been renewed interest in relation to its actions in decreasing reactive oxygen species (Péters et al., 2009). Solutions of this drug are irritant to the tissues.
Butyrophenones
As with phenothiazines, the primary action of butyrophenones is as dopamine antagonists and, in humans, they were classed as major tranquillizers (neuroleptics), and currently as antipsychotics. Used alone, they can cause very unpleasant side effects, including hallucinations, mental agitation, and feelings of aggression. These side effects often are not obvious to an observer and only become known when a human patient recovers from the drug and complains. The incidence is dose related and increases with increased dose rate. Overdose results in dystonic reactions. We do not know whether the subjective effects produced in animals are similar to those that occur in humans, but the unpredictable aggressive behaviour occasionally observed in animals given a butyrophenone suggests they may be.
Cardiovascular and respiratory effects of the butyrophenones are minimal, although arterial hypotension may result from α1-adrenergic blockade. They are potent antiemetics, acting on the chemoemetic trigger zone to prevent drug-induced vomiting, such as may be caused by opioid analgesics, thus making them the drug of choice for the neuroleptic component of neuroleptanalgesia (Brown et al., 2011). Some texts even categorize butyrophenones as injectable anaesthetic agents (Reves et al., 2010), although they can only contribute to the anaesthetic state when in combination with other agents.
In veterinary medicine, drugs in this group are used as sedatives, in neuroleptanalgesic combinations, and may be a useful part of other anaesthetic combinations.
Azaperone
Azaperone, 4′-fluoro-4 [4-(2 pyridyl)-1-piperazinyl] butyrophenone, is a drug licensed in Europe in pigs where its IM administration produces a good, dose-related sedative effect up to the maximum recommended dose for clinical use (4 mg/kg). Pigs may show excitement during the first 20 minutes following injection, particularly if disturbed during this period. Intravenous use of this drug frequently results in a vigorous excitement phase.
Azaperone in clinical doses has minimal effects on respiration; such effect as there is being that of slight stimulation. Clarke (1969) reported a consistent small fall in arterial blood pressure in pigs following the IM injection of 0.3–3.5 mg/kg of the drug. Reductions in cardiac output and heart rate are clinically insignificant (MacKenzie & Snow, 1977).
Azaperone is used both as a sedative and as a preanaesthetic medicant in pigs. It is also used on the farm to sedate pigs before transportation, and to prevent fighting following the mixing of calves or pigs in one pen. When azaperone is used to assist in vaginal delivery of piglets or for caesarean section, the piglets may appear sleepy for some hours after delivery. However, provided they are kept warm they breathe well.
Use in horses, although documented, is not advisable. Doses of 0.4–0.8 mg/kg IM sometimes give good sedation, but some horses develop muscle tremors and sweat profusely (Lees & Serrano, 1976). Intravenous use frequently results in extreme excitement which can cause injury to the horse and/or its handlers (Dodman & Waterman, 1979). The fact that pigs show a similar reaction, coupled with the known central nervous effect in humans, suggests that the excitement is due to a direct effect of the drug.
Droperidol
Droperidol is a potent neuroleptic agent, which is an extremely effective antiemetic and is said to antagonize the respiratory depressant effects of morphine-like compounds by increasing the sensitivity of the respiratory centre to carbon dioxide. Although it was claimed that extrapyramidal side effects were rare, they are produced by overdosing but are sometimes delayed for up to 24 hours after administration of the drug. In humans, droperidol has recently had a resurgence in popularity for premedication (Sneyd, 2009) because of its effect in reducing postoperative vomiting and for counteracting pruritus induced by epidural injection of morphine (Horta et al., 2006). In veterinary medicine, droperidol has been most frequently used in combination, although it has also been used as a sedative in pigs.
Fluanisone
This is 4′-fluoro-4-[4-(o-methoxy)phenyl]-1-piperazinyl) butyrophenone. It is used in the neuroleptanalgesic combination ‘Hypnorm’ (p. 93).
Benzodiazepines
Chlordiazepoxide was first introduced in 1955 and since that time drugs of the benzodiazepine group have been widely used in human and veterinary medicine. Many different compounds now exist differing primarily in bioavailability, permissible route of administration and duration of action. Drugs of this group are utilized to provide:
Benzodiazepine compounds exert their main sedative effects through depression of the limbic system, and their muscle relaxing properties through inhibition of the internuncial neurons at spinal levels. They act selectively at GABAA receptors within the central nervous system. They do not activate the receptor directly, but enhance the response to GABA by increasing the frequency of Cl− channel opening and, therefore, enhance the potentiation of the neural inhibition that is mediated by this transmitter (Olkkola & Ahonen, 2008). The effect of benzodiazepines can be antagonized by inverse agonists or competitive antagonists such as flumazenil. The point of action within the GABAA receptor is not identical to that at which general anaesthetic agents such as thiopental are thought to exert part of their effect (see Chapter 1). Knowledge of the molecular structure of the GABAA receptor and its subunits is advancing rapidly and is beyond the scope of this book, but order of arrangement of ‘subunits’ appears to be responsible for different facets of the overall action (at least in transgenic rats) leading to the future possibility of ‘designer’ drugs with more specific actions (Sneyd & Rigby-Jones, 2010).
It is impossible to induce anaesthesia with benzodiazepine drugs alone in fit healthy animals (Lees, 1979). The benzodiazepines are generally employed in combination with other central nervous depressant drugs to produce anaesthesia and to counteract the convulsant and hallucinatory properties of ketamine and tiletamine. Used for premedication, they improve the quality of induction of anaesthesia and reduce the dose required of subsequent anaesthetic agents. Benzodiazepines cause minimal cardiovascular and respiratory depression, although they may add to the depressant effects of the other anaesthetics.
Many benzodiazepines can be given by the IM, IV, transmucous membrane, oral and rectal routes. Maximal effects may not be apparent for several minutes following IV administration and there are marked differences between individuals in sensitivity. Metabolism is in the liver and, in many instances, metabolites are as active or more active than the parent compound; actions therefore tend to be prolonged.
In a variety of animals, benzodiazepines have the property of stimulating appetite (Van Miert et al., 1989). Diazepam has been particularly widely used for this in cats showing anorexia following illness.
Of the available benzodiazepine drugs, diazepam, midazolam, climazolam and zolazepam have been most utilized in veterinary anaesthesia.
Diazepam
Diazepam is insoluble in water and solutions for injections contain solvents such as propylene glycol, ethanol, and sodium benzoate in benzoic acid. Intravenous injection of many preparations causes thrombophlebitis due to these solvents. An emulsion preparation is non-irritant to veins and is less painful to inject. Diazepam is highly protein bound, and is metabolized by the liver, many of the metabolites being active. In humans, half-life of elimination has been reported as 43 hours. In contrast, in the dog it is shorter at 3.2 hours (Loscher & Frey, 1981) but an active metabolite is longer lasting. In horses, Muir et al. (1982) reported half-lives between 2.52 and 21.6 hours. At clinical dosage rates, diazepam has no significant effect on the circulation or respiratory activity but does produce some muscular relaxation. It has very low toxicity and large doses given to dogs for prolonged periods do not produce changes in metabolic function.
Diazepam has a major role in veterinary practice in the control of convulsions of any origin. However, as with most benzodiazepines, the sedative and hypnotic effects of diazepam alone appear to be minimal and in fit, healthy dogs attempts to use it alone for hypnosis have been unsuccessful. In horses, Muir et al. (1982) did find that at doses exceeding 0.2 mg/kg IV there was some sedation lasting for around 2 hours, but this was accompanied by muscle weakness. This dose of 0.2 mg/kg is used to sedate foals, which will tend to lie down.
The major anaesthetic use of diazepam is a part of an anaesthetic combination protocol, either as premedication, or in combination with other agents. In sick animals (which are more likely to sedate), it reduces the dose of anaesthetic agents subsequently required and its combination with opioids for induction of anaesthesia provides good cardiovascular stability (Psatha et al., 2011). It is particularly useful prior to or in combination with ketamine as it appears to reduce the hallucinations, which seem to be associated with this dissociative anaesthetic agent, and it has been used in this combination in very many species.
Midazolam
Midazolam, (8-chloro-6 (2-flurophenol)-1–methyl-4H imidazo (1,5-a) (1,4)) benzodiazepine is a water-soluble compound yielding a solution with a pH of 3.5. Above pH values of 4.0, the chemical configuration of the molecule changes so that it becomes lipid soluble. The aqueous solution is not painful on IV injection and does not cause thrombophlebitis. Midazolam is metabolized in the liver. In humans, its half-life of elimination is considerably shorter than that of diazepam thus it is less cumulative and recovery is more rapid. In dogs, half-life of elimination has been measured at 77–98 minutes (Hall et al., 1988; Court & Greenblatt, 1992), also shorter than that of diazepam. Midazolam has good biovailability by non-IV routes including oral and transmucous membrane. In humans, it is used to treat seizures, for premedication, as an infusion for sedation in intensive care, and as an IV injection to induce deep sedation and often, amnesia. The dose required in humans is approximately half to one third that of diazepam (Court & Greenblatt, 1992) and the onset of action is slower than a circulation time. In contrast to humans, in adult fit healthy animals, it is difficult to induce sedation with IV midazolam alone, and aberrant excitement reactions can occur (Covey-Crump & Murison, 2008). However, in sick animals, or when combined with agents such as opioids or ketamine, good hypnosis can be achieved, and midazolam, in combination with other agents, now has a major role in the anaesthesia of all species.
The lack of guaranteed sedation with midazolam alone means that there are few animal studies on its use as a sole agent but there are very many publications detailing its use in combination with other analgesic/anaesthetic agents for sedation, as a premedicant and for anaesthesia. This wide literature provides a basis for the recommendations that will be given in the chapters on the individual species.
Climazolam
Climazolam is a potent benzodiazepine which, following IV administration, has a very rapid onset of effect. It has a marketing authorization in Switzerland for use in dogs, but has been used in a wide variety of animals including cattle, sheep, horses and dogs (Rehm & Schatzmann, 1984). It is used as part of anaesthetic combinations. It is particularly effective for use with ketamine and, in horses, this combination has been used by constant rate infusion (CRI) to produce total intravenous anaesthesia (TIVA), the climazolam being reversed with sarmazenil (see below) at the end of surgery to produce a rapid recovery (Bettschart-Wolfensberger et al., 1996). Climazolam (1.0–1.5 mg/kg) has also been used in combination with fentanyl (0.005–0.015 mg/kg) for anaesthesia in the dog (Erhardt et al., 1986).
Zolazepam
This drug is used in animals combined, in a fixed ratio, with the dissociative agent, tiletamine (see Chapter 6).
Benzodiazepine antagonists
Flumazenil
Flumazenil is a potent and specific competitive benzodiazepine antagonist and, in human medical practice, is now being widely employed to reverse midazolam sedation in ‘day case’ patients.
Flumazenil has been reported to reverse diazepam or climazolam sedation in sheep and cattle (Rhem & Schatzmann, 1984) and has also been used in combination with naloxone to reverse climazolam/fentanyl combination anaesthesia (Erhardt et al., 1986). Although to date the veterinary use of flumazenil has been limited by cost, it may be employed in any situation where it may become necessary to reverse the effects of a benzodiazepine drug (Heniff et al., 1997; Wismer, 2002).
Sarmazenil
This antagonist has marketing authorization in Switzerland for use in dogs and cats. It has been used to reverse the effects of climazolam in a number of species (Rehm & Schatzmann, 1984; Bettschart-Wolfensberger et al., 1996). Midazolam-induced ataxia in a horse was effectively reversed with sarmazenil (author KC).
α2-Adrenoceptor agonists
Xylazine has been used as a sedative in animals since 1968 but, at that time, the mechanisms of its complex actions and side effects were not understood. When it was described as ‘both excitatory and inhibitory of adrenergic and cholinergic neurons’ (Kronberg et al., 1966), this statement appeared more than a little confusing. A similar drug, clonidine, was originally used in humans for its powers of local peripheral vasoconstriction but now is used as an antihypertensive and also, off label, as a sedative and analgesic.
These actions and the correctness of the above description only became explicable when Langer (1974) suggested the existence of receptor sites situated presynaptically on the noradrenergic neurons which, when stimulated by noradrenaline, inhibited the further release of this transmitter, thus forming a negative feedback mechanism. Langer suggested further that these presynaptic inhibitory receptors differed from the previously recognized α-adrenoceptors and should therefore be termed α2-adrenoceptors. There are postsynaptic and presynaptic α2-adrenoceptors in both central and peripheral sites. The distinction between α1- and α2-adrenoceptors is made on sensitivity to specific agonist and antagonists. Epinephrine (adrenaline) and norepinephrine (noradrenaline) stimulate both types. For α1-adrenoceptors, phenylephrine and methoxamine are considered to be fairly specific agonists and prazosin to be a specific antagonist. Classically, for pharmacological tests, clonidine was considered a specific α2-agonist and yohimbine an antagonist. Current research utilizes newer and more selective agents.
Norepinephrine is the natural ligand for both α1– and α2-adrenoceptors. When norepinephrine is released from a ‘sympathetic ‘neuron, it acts on both these receptors at the effector site, resulting in ‘sympathetic’ stimulation. However, there are α2-adrenoceptors on the sympathetic neuron which, when stimulated by the released norepinephrine, exert a negative feedback and prevent further release of norepinephrine (Fig. 4.1). Simplistically, the α2-adrenoceptor agonists (α2-agonists) exert their central effects by stimulating these presynaptic α2-receptors, thus preventing the release of norepinephrine and damping or preventing sympathetic drive from the central nervous system.
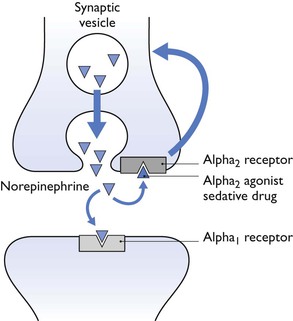
Figure 4.1 This represents a noradrenergic neuron in the central nervous system (CNS). Norepinephrine is the natural ligand and acts at the effector sites (α1– and, peripherally, also α2-receptors). However, in the central nervous system, there are α2-receptors on the noradrenergic neuron; stimulation of these acts as a negative feedback system and prevents further release of norepinephrine. The α2-agonist sedative drugs act at this point, and therefore block release of norepinephrine.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


