Chapter 5
Analgesia
Kate Borer-Weir
Intravenous regional analgesia (IVRA)
Epidural and intrathecal analgesia
Systemic administration of lidocaine
It is difficult to define pain satisfactorily. Everyone has their own idea of ‘what pain is’ but it can be hard to put this experience into words. The International Association for the Study of Pain (IASP) has the following definition: ‘Pain is an unpleasant sensory and emotional experience associated with actual or potential tissue damage or described in terms of such damage.’ This definition relates to human experience and may not be accurate when applied to animals, as they cannot describe emotions brought about by pain. However, the IASP also recommends that ‘the inability to communicate verbally does not negate the possibility that an individual is experiencing pain and is in need of appropriate pain-relieving treatment’. It is now generally accepted that animals feel pain in the same way as humans, as the physiological mechanisms are similar. Controversy exists about whether neonatal animals and fetuses in utero feel pain and which procedures are acceptable to perform on neonates of what age without analgesia.
Physiology of pain
Nociception describes the sensory processes by which noxious stimuli are transmitted to the brain. Stimulation of peripheral nerve endings (nociceptors) generates action potentials in sensory nerves. Different nociceptors respond to different stimuli: mechanoreceptors respond to pressure and stretch; thermoreceptors to heat and cold; and chemoreceptors to inflammatory mediators such as H+, K+ and prostaglandins. Nociceptive impulses are transmitted in Aδ and C fibres. Aδ fibres are small myelinated fibres which transmit the sensation of fast, sharp pain. C fibres are the smallest unmyelinated fibres which detect slow, dull pain. Sensory neurons enter the spinal cord via the dorsal horn (ending preferentially on cells in laminae 1, 2 and 5) where they may stimulate a simple withdrawal reflex, passing the information directly to motor nerves to cause movement of a limb away from a painful surface for example.
Concurrently, information passes up the spinal cord in pathways including the spinothalamic and spinoreticular tracts (in the ventrolateral white matter), through many interneurons to reach the brain, particularly the thalamus. From here, information passes to higher centres in the cerebral cortex.
In the spinal cord, many receptors and mediators modulate the information passing to the brain. Excitatory transmitters, including glutamate, substance P and prostaglandins, propagate the pain impulse. Inhibitory transmitters, such as endorphins, norepinephrine, serotonin (5-hydroxytryptamine; 5-HT) and γ-aminobutyric acid (GABA), reduce the painful stimuli reaching the brain. This forms the basis of the ‘gate theory’, an adaptive method of controlling the painful stimuli which reach the brain. It evolved to allow escape from life-threatening situations and means that the brain does not perceive the true amount of pain until the concentrations of inhibitory neurotransmitters, such as epinephrine and norepinephrine, drop sufficiently to allow nociceptive transmission to resume. Descending pathways from the brain (particularly the periaqueductal grey; PAG) also contribute to this inhibitory control. Stimulation of non-nociceptive neurons (e.g. by touch or pressure) can inhibit transmission in nociceptive neurons.
The nervous system demonstrates ‘functional plasticity’ and numerous changes occur after continued nociceptive stimulation. The products of tissue breakdown and damage have direct and indirect effects on sensory neurons. Neurotransmitters are released in the periphery from nociceptive nerve terminals, including substance P, bradykinin, neurokinin A and calcitonin gene-related peptide (CGRP). These substances cause vasodilation, extravasation of plasma proteins, stimulation of inflammatory cells to release more mediators and excitation of other sensory and sympathetic neurons. This produces an ‘inflammatory soup’, sensitizing nociceptors and sensory nerves causing peripheral sensitisation (primary hyperalgesia: increased sensitivity to painful stimulation at the site of injury). Prostaglandins in particular increase the sensitivity to pain by lowering the threshold at which nociceptive neurons fire. Other changes in the peripheral nervous system include spontaneous, ectopic activity in primary afferent nerves and sprouting of sympathetic nerves around the dorsal root ganglion.
Glutamate, one of the primary excitatory neurotransmitters in the CNS usually acts via AMPA (α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid) and neurokinin receptors. However, continued nociceptive stimulation within the spinal cord releases glutamate and substance P and removes the Mg2+ plug which usually blocks the N-methyl-d-aspartate (NMDA) receptor for glutamate, allowing priming of this receptor. Once the NMDA receptor is active, glutamate binding allows Ca2+ entry and stimulates second messenger release. Continued nociceptive stimulation alters the distribution of AMPA receptors within the dorsal horn and there may be loss of inhibitory neurons containing GABA. These changes within the CNS are known as central sensitization (secondary hyperalgesia), representing the spread of pain sensitivity (a lowered threshold to pain) outside the area of the original injury. The development of central sensitization may be regulated by the c-fos gene, which is expressed rapidly in spinal and supraspinal neurons following nociceptive stimulation.
Allodynia describes altered pain perception, whereby non-painful stimuli, such as touch, are perceived as painful by the patient. It commonly occurs during neuropathic or chronic pain and can be very hard to control with analgesic drugs. The mechanism behind allodynia may include sprouting of non-nociceptive A fibres into areas of the dorsal horn where C fibres normally terminate.
Optimal pain management should include preventing the development of chronic or neuropathic pain and the associated changes in the nervous system associated with hyperalgesia. Pre-emptive analgesia describes the provision of analgesia before any painful stimuli have occurred. This prevents the development of hypersensitization during surgery and should result in less postoperative pain that is easier to manage with lower doses of analgesic drugs. In animals with pre-existing injuries, such as those with orthopaedic problems or after road traffic accidents, provision of analgesia as soon as possible may reduce the development of sensitization but not completely eliminate it. There are conflicting reports on the benefits of pre-emptive analgesia and differences between experimental and clinical studies. Meta-analyses in human medicine have demonstrated both a positive effect (Ong et al., 2005) and no benefit (Moiniche et al., 2002).
The goal of multimodal analgesia is to block the pain pathways at multiple sites, using agents with different modes of action, thus providing more effective pain control with decreased dose rates and reduced prevalence or severity of toxic side effects. Drugs can be used to inhibit transduction and peripheral sensitization, inhibit transmission of sensory impulses, modulate the spinal pathways involved in central sensitization and to inhibit perception of a noxious stimulus in the brain.
In humans, analgesia is considered to be effective when the clinical signs of pain disappear. Similarly, a drug may be considered to be analgesic in an animal if signs associated with pain are ameliorated following administration. It is important to be sure that the drug effect is truly analgesia and not merely preventing the animal displaying the behavioural signs of pain. An extreme example would be administration of a neuromuscular blocking drug that causes paralysis without affecting the perception of a painful stimulus.
Pain assessment
Pain assessment is a crucial component of effective analgesia. Clinical signs of pain (Box 5.1) are unreliable, non-specific, and differ between species. Heart rate, respiratory rate and pupil dilation were not useful indicators of pain in dogs recovering from surgery (Holton et al., 1998a). Furthermore, these parameters were poorly correlated with subjective pain scores in dogs after orthopaedic surgery (Conzemius et al., 1997). In horses following orthopaedic surgery, measurement of blood pressure, but not heart rate, was correlated with behavioural assessment of pain (Bussieres et al., 2008).
Pain assessment often relies on behavioural indicators of pain (see Box 5.1), but these vary between species, breeds, and individuals. Behaviour is also modified when the animal is in a hospital environment. Behavioural signs of pain depend on the origin of the pain, for example, visceral or somatic pain, and the type of procedure performed. Much of the early research on pain assessment was performed on dogs in the acute postoperative period. Now research includes behaviours associated with chronic pain (including cancer), medical pain and pain in domestic, agricultural, and exotic species. Behavioural assessment of pain in horses and donkeys (Ashley et al., 2005) and laboratory animals (Roughan & Flecknell, 2004; Roughan et al., 2009) have been reviewed.
Measurement of ‘stress hormones’ including epinephrine, cortisol, endorphins, glucose and insulin have not proved useful in diagnosing pain in individual animals in the clinical setting, although they can be supportive in research studies to distinguish between treatment and control groups. They are increased by conditions causing stress such as hospitalization and general anaesthesia.
When assessing behaviour, the animal should first be observed undisturbed and then during a series of interactions. Some behaviours arising from poor general health, medical problems, anxiety or fear may be mistakenly assessed as pain behaviour, and other behaviours such as aggression may be normal for the individual. Pain may also be evident through the suppression of normal behaviour, rather than additional pain-related behaviour. Prey animals in particular are unlikely to display overt pain behaviours as an evolutionary survival mechanism, and pain assessment in these animals can be difficult. Pain behaviours are useful to detect the presence of pain but they may not indicate the severity of pain.
Pain scales
Pain scales were created to increase objectivity in pain assessment and to decrease variability between scorers and animals. Pain scales are not infallible but use of a pain score helps to monitor changes in the same animal over time, providing information for adjustments in the analgesic regimen. During preanaesthetic evaluation, the patient should be assessed for pre-existing pain and a judgement made regarding the degree of pain likely to be experienced during the procedure. For example, minor procedures such as laceration repair would be expected to cause less pain than major orthopaedic surgery or thoracotomy.
A simple descriptive score (SDS) uses a scale from 0 to 3 to score no pain (0), mild pain (1), moderate pain (2) and severe pain (3). This is simple and quick to perform during a busy working day but is subjective and relatively imprecise. Inter-observer variability using an SDS was fair for dogs post-surgery assessed by three or four vets (Holton et al., 1998b).
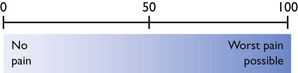
The visual analogue scale (VAS) is a line 100 mm long anchored at either end with 0 = no pain and 100 = worst pain possible (Fig. 5.1). The observer places a mark on the line at their assessment of the patient’s pain and the distance from 0 to the mark is measured. There is considerable inter-individual variability in the score assigned by different observers using a VAS (Holton et al., 1998b), but a single observer can become consistent with practice.
A simple numerical rating scale (NRS) can be used to assign a score from 0 to 10 for the assessed degree of pain. This score is commonly used to assess equine lameness. The numbers do not have specific descriptors, and there is often considerable inter-observer variability in scoring. A more involved NRS uses multiple categories containing specific descriptors of different types of behaviour, each of which has a numerical score. One descriptor from each category is selected and the total score from each category added to give an overall pain score. This gives a more thorough pain evaluation, but is also more time consuming. It can be limiting if the behaviour displayed by the individual animal does not match exactly any of the descriptors in the scale. An example is the University of Melbourne Pain Scale (UMPS), which includes both behavioural responses and physiological data (Firth & Haldane, 1999). A similar composite pain scale based on the NRS has been developed to assess orthopaedic pain in horses (Bussieres et al., 2008).
The previous pain scores are all unidimensional, assessing only pain intensity. However, it is well known from human experience that pain has additional significant sensory and affective qualities. These are addressed in multidimensional pain scores. The McGill pain questionnaire uses a series of descriptive words, categorized according to their sensory, affective or evaluative capacity, each of which is anchored to a numerical score to assess pain in humans (Melzack, 2005). The Glasgow Composite Pain Tool was developed in a similar way to assess acute surgical pain in dogs (Holton et al., 2001). A series of expressions and descriptions of behaviour were reduced to specific words and phrases to describe different aspects of the pain experience. The aim was to reduce inter-observer variability and bias. The Short Form of the Glasgow Pain Scale is an extension of this, incorporating a numerical score to aid clinical use (Reid et al., 2007). Many of the problems associated with pain assessment in animals and limitations of some of the scores currently available, including validation of the score and linearity of the scale used, have been reviewed (Flecknell & Roughan, 2004).
Analgesiometry systems (nociceptive threshold testing) are commonly used in the initial testing of analgesic drugs to assess relative potency, onset and duration of action. These involve the use of a controlled and repeatable stimulus, often a pressure or thermal stimulus, applied before and after administration of an analgesic (Dixon et al., 2002). The animal is observed for a specific aversion response, such as turning of the head, at which point the stimulus is stopped and the maximum value noted. This provides a very useful starting point for the comparison of different analgesics in animals, but it must be appreciated that clinical pain is much more complex and the performance of a drug in an analgesiometry system may not be representative of its efficacy as an analgesic in different clinical pain scenarios (Melzack, 2005).
Specific pain scores for different species are discussed further in the relevant species chapters.
Non-steroidal anti-inflammatory drugs (NSAIDs)
NSAIDS have anti-inflammatory, analgesic and antipyretic actions. They are among the most commonly used drugs in veterinary medicine to relieve pain and inflammation in all species. Many can be given initially by injection and then prescribed orally for longer-term use. They are not subject to controlled drugs legislation and there are many different agents licensed for use in different species and in different countries. Salicylate is a naturally occurring NSAID found in willow which has been used for thousands of years. However, the first synthetic NSAID was synthesized in the 1870s (Lees et al., 2004b). Acetylsalicylate (acetylsalicylic acid; aspirin) still has an annual consumption of approximately 80 billion tablets per year.
Pharmacology
NSAIDS inhibit production of prostaglandins (PGs) and thromboxane (TX) via inhibition of cyclo-oxygenase (COX; Fig. 5.2). PGs, TX and leukotrienes (LTs) are generated from arachidonic acid which is a 20-carbon fatty acid, released from cell membrane phospholipids after cell damage. The conversion of cell membrane phospholipid to arachidonic acid is catalysed by phospholipase A2, which is inhibited by glucocorticoids (Fig. 5.2).
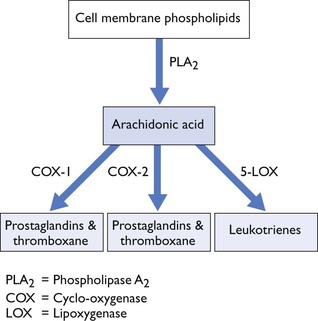
Figure 5.2 Metabolism of phospholipids to form arachidonic acid, prostaglandins, thromboxane and leukotrienes.
PGs have many homeostatic functions in the body, including maintenance of the gastric mucosal barrier and protection against gastric ulceration. PGs also help to maintain adequate renal blood flow, especially in situations where this is reduced. As well as their physiological functions, PGs are key inflammatory mediators, acting synergistically with other mediators, such as histamine and bradykinin, to increase pain, capillary permeability and oedema.
In general, NSAIDs are well absorbed after oral administration, are highly protein bound in plasma and have a large volume of distribution (Lees et al., 2004b; Curry et al., 2005). There are significant species differences in pharmacokinetics, including clearance, half-life and COX selectivity, meaning that extrapolation between different species is not recommended (Lees et al., 2004b). Genetic polymorphisms in response to NSAIDS exist, for example clearance and elimination half-life of celecoxib in Beagles differed by more than twofold in animals with hepatic microsomal enzyme differences (Paulson et al., 1999). Disease states may also reduce clearance and thus increase the effective half-life of NSAIDS (Lohuis et al. 1991). The pharmacokinetics can also differ depending on whether NSAIDs are administered before or after general anaesthesia (Lascelles et al., 1998).
COX-1 and COX-2
Two iso-enzymes of COX with slightly different chemical structures were discovered in 1991. Initially, these two isoforms were thought to have quite separate and different roles. COX-1 was regarded as the constitutive enzyme, responsible for the production of PGs with physiological or ‘housekeeping’ functions including gastric and renal protection. COX-2 was thought to be the inducible enzyme, found at sites of injury producing PGs with inflammatory functions.
Classical NSAIDs inhibit both COX-1 and COX-2 with COX-1 inhibition resulting primarily in the side effects of these drugs including gastric ulceration and renal failure. Drug manufacturers sought to develop NSAIDs with COX-2 selective inhibitory activity. They hoped this would remove the risk of side effects, but retain the beneficial anti-inflammatory effects.
NSAIDs are thus classified into:
•Non-selective drugs which inhibit both COX-1 and COX-2 equally (classical NSAIDs)
•COX-2 selective which selectively or preferentially inhibit COX-2
•COX-1 selective which selectively or preferentially inhibit COX-1
Coxib drugs are large molecules which fit into the binding pocket of the COX-2 receptor, but are too large to fit into the smaller COX-1 receptor, resulting in their selectivity (Bergh & Budsberg, 2005).
The COX-1:COX-2 ratio describes the selectivity of the NSAID and refers to the dose of drug which must be given to inhibit each of the two isoforms. Non-selective agents have a COX-1:COX-2 ratio close to 1. COX-2 selective inhibitors have a ratio greater than 1 (i.e. a higher dose of drug must be given to inhibit COX-1 compared to the dose required to inhibit COX-2). The specific COX-2 inhibitors typically have a COX-1:COX-2 ratio of 100 or more. This means you would need to give 100× more drug to inhibit COX-1 than you would to inhibit COX-2.
The COX-1:COX-2 ratio varies depending on the type of assay used. The whole blood assay is considered the most applicable to the in vivo situation. Ratios also vary between different tissues and species. For example, in the dog, carprofen is thought to be COX-2 selective; in the horse, it is non-selective, whereas in humans, it is COX-1 selective (Brideau et al., 2001; Lees et al., 2002, 2004b). Many COX-1:COX-2 ratios are based on the IC50 for each isoform (the concentration of drug giving 50% inhibition of COX). However, it is more relevant to look at the ratio that reflects the clinically desirable degree of inhibition for each enzyme. This is believed to be 20% or less for COX-1 inhibition and 80% or more for COX-2 inhibition, resulting in an IC20 COX-1:IC80 COX-2 ratio. This ratio is often quite different from the IC50 ratio for a given drug.
Initially, the roles of COX-1 and COX-2 were thought to be quite separate with little overlap in the functions of the PGs produced by each isoform. However, we now know that there is a considerable overlap between the two (Wallace, 1999; Curry et al., 2005). In particular, COX-1 is upregulated at sites of injury and catalyses the production of inflammatory PGs. COX-1 is involved in spinal cord pain processing and hypersensitization, and COX-1 inhibition in the spinal cord reduces postoperative pain in rats (Zhu et al., 2005). COX-2 has physiological roles in the body, including gastric ulcer healing and it is constitutively expressed in many tissues, including the kidney, brain, reproductive tract and ciliary body of the eye. The role of COX-2 in different tissues and the effects of COX-2 inhibition have been reviewed (Bergh & Budsberg, 2005). Therefore, selective COX-2 inhibition does not completely eliminate the side effects associated with classical NSAIDS.
There is continuing debate over the presence of a third isoform of COX, COX-3, within the CNS (Kis et al., 2005). Some consider this to be a separate isoform, others feel that it is merely a splice variant of COX-1 and should not be considered separately from COX-1, while others question its existence at all (Kis et al., 2005). COX-3 was first described in dogs where it was primarily found in the cerebral cortex and was produced by the COX-1 gene (Chandrasekharan et al., 2002). Others have reported that it has a low expression level, limiting its clinical importance (Kis et al., 2005). Some NSAIDs may have a central component to their analgesic actions mediated by COX-3. Most NSAIDs cross the blood–brain barrier poorly, with the exception of paracetamol, which appears to exert its main antipyretic and analgesic effects in the CNS. Paracetamol has minimal inhibitory effects on peripheral COX activity in vitro, but produces analgesia in different animal models of pain and nociception (Warner & Mitchell, 2002). It also lacks ulcerogenic potential in the gastrointestinal tract. It may be a potent inhibitor of COX-3 (Chandrasekharan et al., 2002) with weak activity against COX-1 and COX-2. However, in humans, the existence of COX-3 has not been proven and, as a result, interest and research in the area has waned. The target site of action of paracetamol is still unclear. On a similar note, some studies have reported that carprofen is only a weak inhibitor of COX in the periphery (Lees et al., 2002), yet it is a powerful analgesic agent. It has been hypothesized that central inhibition of COX-3 may contribute to carprofen’s analgesic actions.
Other studies have hypothesized that the central analgesic effect of NSAIDs is mediated by activation of descending inhibitory pathways (both opioidergic and adrenergic) within the spinal cord (Lizarraga & Chambers, 2006).
NSAIDs may also exert anti-inflammatory actions through a variety of COX-independent mechanisms, including key signaling pathways involving phosphatidylinositol 3-kinase (PI3K), peroxisome proliferator-activated receptors (PPAR), nuclear factor kappa B (NFκB) and mitogen-activated protein kinases (MAPK) among others. These mechanisms have been recently reviewed (Little et al., 2007b; KuKanich et al., 2012).
Side effects
The side effects of NSAIDs are mainly related to undesirable inhibition of COX-1 and are generally common to the group although the likelihood of each varies slightly depending on the COX-1:COX-2 selectivity ratio of the specific drug and the particular species concerned. Gastrointestinal ulceration is one of the commonest side effects in both humans and animals. PGs increase blood flow to the gastric mucosa and also increase secretion of protective mucous and bicarbonate. COX-2 is upregulated around existing gastric ulcers and this isoenzyme may play a key role in ulcer healing. In humans and small animals, gastric lesions and gastric ulcerations are commonest. In horses, while gastric ulceration occurs in response to NSAIDs, lesions involving the colon resulting in right dorsal colitis may also occur, particularly after an NSAID overdose. Combination therapy with two different NSAIDs or administration above the recommended dose also increases the risk of serious GI side effects (Reed et al., 2006).
Both carprofen and meloxicam had minimal effects on gastrointestinal permeability in healthy dogs, however, individual variation was apparent, with permeability increasing in some (Craven et al., 2007), while gastric lesions on endoscopy and occult blood on faecal examination have been reported in experimental dogs (Luna et al., 2007). In horses, non-selective COX inhibitors such as flunixin slow the recovery of ischaemic-damaged intestinal mucosa (Campbell & Blikslager, 2000; Tomlinson & Blikslager, 2004), mainly by affecting the integrity of tight junctions between intestinal cells and thus increasing paracellular permeability. This effect was not seen with meloxicam, a COX-2 selective inhibitor in the horse (Little et al., 2007a).
COX-inhibiting nitric oxide donors (CINODs) are nitrosoesters of classical NSAIDs, which may have a better GI safety profile than NSAIDS. In the body, CINODs are broken down to release the parent NSAID and nitric oxide (NO). NO is a vasodilator which may help to prevent local ischaemia in the GI tract associated with COX inhibition or, alternatively, NO may prevent neutrophil attachment to gastric mucosal blood vessels. The pharmacology of the first CINOD to be developed, naproxcinod, has been reviewed (Wallace et al., 2009), although there have been no reports of the use of CINODs in veterinary medicine.
In the kidney, PGs are released from the macula densa in response to vasoconstriction of the afferent arteriole. The resultant vasodilation helps to maintain adequate renal blood flow and glomerular filtration rate (GFR). Adverse renal effects of NSAIDS are most likely to occur during periods when renal blood flow is already compromised, such as hypotension due to haemorrhage, shock or general anaesthesia. These effects appear to be similar with both non-selective NSAIDs and selective COX-2 inhibitors as both COX-1 and COX-2 are constitutively expressed in the kidney. Perioperative carprofen had no effect on GFR or renal blood flow in experimental, healthy anaesthetized Beagles (Frendin et al., 2006) and meloxicam had no significant effect on renal function in otherwise healthy dogs rendered hypotensive under anaesthesia (Bostrom et al., 2006). NSAID-induced nephrotoxicity in humans is more likely in patients also receiving diuretics and/or ACE inhibitors (Weir, 2002).
The different COX isoforms are responsible for producing PGs and TX with different vascular effects. Prostacyclin (PGI2) is produced under the action of COX-2 in the vascular endothelium. PGI2 inhibits platelet aggregation, causes vasodilation and inhibits the proliferation of vascular smooth muscle. Thromboxane A2 (TXA2) is produced by COX-1 in platelets and causes platelet aggregation, vasoconstriction and proliferation of vascular smooth muscle. Usually, the body is able to maintain equilibrium in the production of PGI2 and TXA2. Aspirin primarily inhibits COX-1, reducing production of TXA2. This can result in bleeding, but is also the reason for the use of low-dose aspirin to prevent thrombosis in susceptible human patients after heart attacks and strokes. Conversely, specific COX-2 inhibitors reduce production of PGI2 and leave the body in a ‘pro-aggregatory’ state, with an increased susceptibility to thrombotic events in humans. The risk of these vascular thrombotic events led to the withdrawal of many coxib drugs from the human market. The thrombotic risk to animals treated with coxib drugs is unclear at the moment, as long-term treatment was necessary in humans before the risk became evident. Animals have a different vascular wall structure to humans and tend to have a lower risk of thrombotic vascular events overall, so there may be minimal risk associated with the use of coxib drugs in veterinary species.
Drugs available
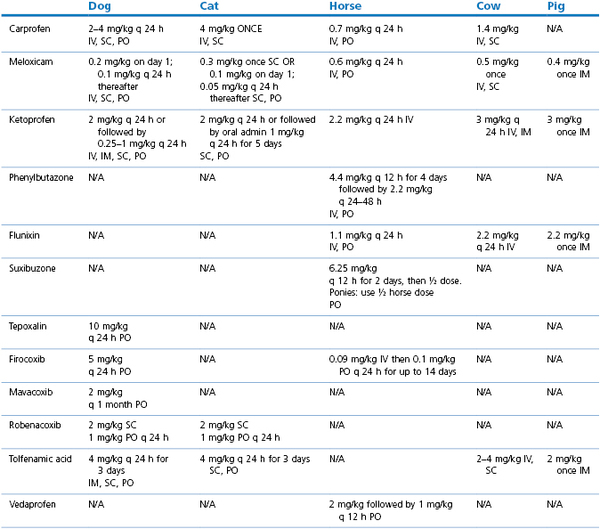
The use of NSAIDs in the cat (Lascelles et al., 2007) and dog (KuKanich et al., 2012) have been comprehensively reviewed. Most clinical studies comparing the analgesic efficacy of different NSAIDs in veterinary species have concluded that there are minor, if any, differences in efficacy between the commonly used drugs (Slingsby & Waterman-Pearson, 2000a, 2002; Deneuche et al., 2004; Laredo et al., 2004; Erkert et al., 2005; Leece et al., 2005). This may reflect true equivalency between different drugs or may be due to limitations in pain assessment scores to detect subtle differences in analgesia in animals. The evidence for long-term use of NSAIDs to treat canine osteoarthritis has recently been reviewed (Innes et al., 2010) with the authors concluding that longer-term treatment had beneficial effects compared to acute treatment, with a low incidence of adverse effects. Drug doses of commonly used NSAIDs licensed in veterinary species in the UK are listed in Table 5.1.
Carprofen
At clinically effective doses, carprofen is a weak inhibitor of COX iso-enzymes in the periphery (Brideau et al., 2001; Lees et al., 2002, 2004a). Its anti-inflammatory actions may be due to a number of different mechanisms, including inhibition of reactive oxygen species and lysosomal enzymes, stimulation of proteoglycan synthesis and modification of the release of cytokines such as IL-1 and IL-6 (Armstrong & Lees, 2002; Lees et al., 2002). Central inhibition of COX-3 has also been proposed to explain carprofen’s analgesic effect. Other studies have shown that carprofen is mildly COX-2 preferential in dogs (Kay-Mugford et al., 2000; Wilson et al., 2004). In cats, the recommended dose of carprofen causes 95% inhibition of COX-2 for 42 hours (Giraudel et al., 2005). The COX-1:COX-2 ratio in the horse is around 1.6–2, making carprofen non-selective or very mildly COX-2 selective (Brideau et al., 2001; Beretta et al., 2005).
Meloxicam
Meloxicam is COX-2 selective (Kay-Mugford et al., 2000). It reduced production of inflammatory mediators in synovial fluid (PGE2, substance P and bradykinin) and reduced activity of matrix metalloprotease (MMP) enzymes and markers of cartilage catabolism such as glycosaminoglycans (de Grauw et al., 2009). Meloxicam administered to dogs for 10 days had no effect on platelet function or blood clotting (Brainard et al., 2007). A possible adverse drug reaction to meloxicam has been reported in a dog which developed cutaneous and ocular lesions 24–48 hours after administration (Niza et al., 2007).
Ketoprofen
Ketoprofen is a non-selective or COX-1 selective NSAID, licensed for use in a variety of species. It is not generally considered a very potent analgesic or anti-inflammatory drug, but has a good antipyretic action.
Phenylbutazone
Phenylbutazone is most commonly used in horses where it is frequently used orally for long-term treatment of chronic musculoskeletal conditions. It is slightly COX-1 selective in the horse, with a COX-1:COX-2 ratio around 0.5 (Beretta et al., 2005).
Suxibuzone
Suxibuzone is a prodrug for phenylbutazone in horses and is rapidly converted to phenylbutazone and oxyphenbutazone after absorption. Suxibuzone and phenylbutazone had comparable efficacy (Sabate et al., 2009), but suxibuzone had lower gastric ulcerogenic potential than phenylbutazone (Monreal et al., 2004). The difference in ulcerogenic potential between the two drugs was hypothesized to be due to a local toxic effect of phenylbutazone in the gastric mucosal cells and unrelated to COX inhibition; however, both drugs were administered at more than twice their respective recommended doses (Monreal et al., 2004).
Flunixin
Flunixin is COX-1 selective in the horse, with a COX-1:COX-2 ratio of 0.4 (Beretta et al., 2005). It is commonly used in the treatment of orthopaedic and soft tissue conditions and is a particularly good analgesic for equine colic. There is concern that flunixin may mask increasing levels of pain indicative of the need for surgical intervention in colic, and it should thus be used with caution until a diagnosis has been reached.
Firocoxib
Firocoxib has an IC50 COX-1:COX-2 selectivity ratio (in a canine whole blood assay) of 384, making it highly COX-2 specific (Brideau et al., 2001; McCann et al., 2004). Although not currently licensed in cats, experimentally it has a COX-1:COX-2 ratio of 58 in this species (McCann et al., 2005). According to the manufacturer’s data, after oral administration, firocoxib is detected in plasma within 15 minutes and peak plasma concentrations are reached within 90 minutes. Plasma concentrations are maintained for 24 hours, making once daily dosing possible. The safety of firocoxib has been evaluated in experimental dogs (Steagall et al., 2007). In dogs with arthritis, firocoxib was as efficacious (Pollmeier et al., 2006) or more efficacious (Hazewinkel et al., 2008) than carprofen.
The pharmacokinetics of firocoxib have been described in the horse (Kvaternick et al., 2007). In horses with arthritis, firocoxib had similar efficacy to phenylbutazone (Doucet et al., 2008). Both firocoxib and flunixin produced effective analgesia in horses after gastrointestinal surgery but, unlike flunixin, firocoxib did not impair gastrointestinal mucosal healing after ischaemia (Cook et al., 2009a).
Robenacoxib
Robenacoxib is a novel specific COX-2 inhibitor licensed for use in dogs and cats (King et al., 2009), producing good anti-inflammatory, analgesic and antipyretic effects in experimentally-induced inflammation in cats (Giraudel et al., 2009). The drug has a short residence time in blood but accumulates in inflammatory exudate (King et al., 2009). Using in vitro whole blood assays, the IC50 COX-1:COX-2 ratio in cats was 502 and in dogs was 129; whereas the more clinically relevant IC20 COX-1:IC80 COX-2 ratio in cats was 17 and in dogs was 20 (Giraudel et al., 2009b; King et al., 2010). The use of 2 mg/kg in cats resulted in 5% inhibition of COX-1 and 90% inhibition of COX-2 over 12 hours (Giraudel et al., 2009b). Bioavailability was 84–88% after SC or PO administration in fasted dogs, but dropped to 62% in fed dogs (Jung et al., 2009).
Mavacoxib
Mavacoxib has been recently marketed in dogs as a long-term oral drug administered once monthly. It has a low rate of elimination in the bile, resulting in a prolonged plasma half-life. The elimination half-life was longer in client-owned dogs (39 days) than experimental Beagles (19 days). Five percent of client-owned dogs had an elimination half-life greater than 80 days, making this subgroup at increased risk of toxic side effects after repeated monthly dosing. Its COX-1:COX-2 ratio is 40, making mavacoxib COX-2 selective, rather than specific. In manufacturer’s data involving a comparison of mavacoxib with carprofen in 600 dogs, the incidence of GI side effects was similar with both drugs.
Dual Inhibitors: tepoxalin
Tepoxalin inhibits both COX iso-enzymes as well as 5-lipoxygenase (5-LOX) resulting in reduced formation of PGs, TX and leukotrienes (LTs). The leukotrienes are important inflammatory mediators in their own right, involved in many different inflammatory, allergic and neoplastic conditions (Goodman et al., 2008) so inhibition of 5-LOX should contribute to the anti-inflammatory and analgesic properties of these drugs (Curry et al., 2005). Leukotrienes cause neutrophil recruitment and activation in the endothelium and the gastric lining, contributing to gastric irritation and ulceration (Moreau et al., 2005). One of the theories about the side effects of NSAIDs is that when COX is inhibited, arachidonic acid is shunted into the 5-LOX pathway, increasing production of leukotrienes and thus increasing the risk of gastric ulceration (Kirchner et al., 1997). The dual inhibitors should avoid this biological imbalance by inhibiting both enzyme pathways.
At present, there is little evidence about the clinical use of tepoxalin in dogs. Most published evidence has been conducted in a laboratory setting using rats (Wallace et al., 1993). Tepoxalin was compared to meloxicam in experimental dogs, although clinical efficacy or side effects were not evaluated (Agnello et al., 2005). Tepoxalin was more effective than carprofen or meloxicam at inhibiting the inflammation (increased PGE2 concentrations) due to experimentally-induced uveitis in dogs (Gilmour & Lehenbauer, 2009). After its initial launch, there were anecdotal reports from vets of gastrointestinal complications (vomiting and diarrhoea) associated with tepoxalin treatment in dogs, in contrast to the experimental GI safety demonstrated with rats. There is still a need for large scale published clinical trials in dogs. In dogs, tepoxalin has a high first pass effect after oral administration resulting in rapid conversion to an acid metabolite (Homer et al., 2005). This acid metabolite is a potent inhibitor of COX. As a result, tepoxalin in dogs only inhibits 5-LOX for a short period of time, whereas the inhibition of COX is much longer lasting, so the drug is actually acting as a non-selective classical NSAID, or even a COX-1 selective drug. This may be the reason for the side effects reported in dogs.
Licofelone is another dual inhibitor which reduces lameness in experimental dogs, although it is not currently commercially available in the UK (Moreau et al., 2005, 2007).
Opioids
Opioids have been used as painkillers in humans for at least 2000 years. Opium is the prototype opioid, derived from the poppy (Papaver somniferum), its major active constituent being morphine. There is now a wide range of both naturally occurring and synthetic opioids available, so that the clinician has an enormous range of choice. They are principally used to provide analgesia but some are cough suppressants. Unfortunately, these drugs have a wide range of side effects and, in humans, they cause euphoria and addiction, rendering them liable to abuse and resulting in controls on their supply and use. Abuse is not a feature of their use in veterinary patients. At least in humans, it seems that the euphoric effects of opioids contribute to their analgesia as patients are unconcerned by any residual pain. Whether this is also true in animals can only remain a speculation.
Pharmacology
Endogenous opioid ligands, such as the endorphins, dynorphins and enkephalins, are found in the CNS. Opioids, both natural and synthetic, bind to opioid receptors (Martin et al., 1976). The main opioid receptors are known as µ (mu, OP3, MOP), κ (kappa, OP2, KOP) and δ (delta, OP1, DOP). Opioid receptor terminology is still the subject of considerable debate, and a unanimous decision has not yet been reached on the naming of the receptors. Initially, Martin et al. (1976) hypothesized that there was also a σ (sigma) receptor, although this is now thought not to be a true opioid receptor. Recently, another receptor has been included in the family: the nociceptin orphanin FQ receptor (N/OFQ, OP4, NOP). Opioid receptors are G-protein coupled receptors causing cellular hyperpolarization due to potassium efflux, closing of calcium channels and reduced cyclic adenosine monophosphate (cAMP) production via inhibition of adenylate cyclase. These cellular effects reduce cell excitability and inhibit release of neurotransmitters.
The µ receptor is found throughout the CNS, particularly in the dorsal horn of the spinal cord and in areas of the brain (such as the periaqueductal grey; PAG) responsible for descending inhibitory control of nociceptive transmission. Binding to the µ receptor is responsible for the majority of opioid-related side effects, including euphoria and addiction in humans. The µ receptor is the main receptor responsible for the analgesic actions of opioids, although there is evidence, at least in humans and rodent models, for some analgesia mediated by the κ receptor, possibly acting in the dorsal horn of the spinal cord. However, analgesia induced by κ agonists is relatively weak and has a ceiling effect. The κ receptor may be responsible for dysphoria. There is some evidence that κ agonists can antagonize some of the side effects induced by µ agonists. The δ receptor may modify the actions of opioids at other receptors. The N/OFQ receptor has a pronociceptive (i.e. antianalgesic) action supraspinally and endogenous N/OFQ may be responsible for setting a pain threshold. The role of N/OFQ in modulation of analgesia is incompletely understood, although N/OFQ receptor antagonists show potential as long-lasting analgesics (Zaveri, 2003).
Drugs may be agonists or antagonists at all opioid receptors, or may exert their actions through one specific type of receptor. Furthermore, some drugs are agonists at one type of receptor and antagonists at another (agonist–antagonists). Partial opioid agonists also exist which exert agonist actions at the specific receptor, although not usually as profound as a pure agonist. Partial agonists often provide sufficient analgesia for clinical use and may also be able to antagonize some of the side effects of pure agonists. Partial agonists are less liable to abuse and so are subject to less stringent controls over their use. They are discussed further under buprenorphine below.
In humans, oral opioids often have reasonably good bioavailability and long elimination half-lives, whereas in dogs, opioids such as methadone and morphine have short elimination half-lives and are rapidly cleared from the circulation. Oral opioids in animals generally have very poor bioavailability due to high first pass metabolism in the liver (KuKanich et al., 2005) making oral administration unfeasible in most veterinary species. Care is necessary when administering systemic opioids to animals with significant hepatic disease as the metabolism of opioids is reduced and thus their half-life extended (Waterman & Kalthum, 1990). There is currently increasing interest in encapsulation of different opioids into liposome formulations to provide sustained release preparations for use in humans, enabling prolongation of dosing intervals.
Opioids have synergistic actions with most sedatives and anaesthetic agents. In general, opioids have an anaesthetic sparing effect (minimum alveolar concentration [MAC] sparing effect; see Chapters 1 and 7) in small animals, allowing lower doses of injectable and inhalational agents to be used, thus reducing the side effects of these agents. In horses, studies have generally failed to find a MAC sparing effect of opioids (Bennett et al., 2004) and this may be related to the central excitatory effects of opioids in horses. However, clinical studies have noted that horses administered opioids as part of an anaesthetic regimen were easier to maintain at a stable plane of anaesthesia and required fewer supplementary doses of injectable anaesthetic agents (Clark et al., 2005).
Side effects
The side effects of opioids vary with species and drug. Side effects are much more common when high doses are given experimentally to pain-free animals, and many side effects are less serious when used clinically in painful animals.
Respiratory depression is commonly cited as the most important side effect of opioids, but this is generally much less of a problem in animals than in humans. High doses of the potent full µ agonists, especially administered IV or in an anaesthetized animal are most likely to cause problematic respiratory depression, necessitating the use of intermittent positive pressure ventilation (IPPV). During the recovery period, or postoperatively, when IPPV cannot be used, highly respiratory depressant opioids are best avoided. Respiratory depression may be more problematic in patients which cannot tolerate even mild increases in PaCO2, such as animals with head trauma or raised intracranial pressure. In animals with thoracic pain, opioids may actually improve respiratory function as a result of their analgesic effect. The cough reflex is depressed and this may increase the risk of aspiration.
In dogs, opioids usually produce sedation. However, historically, there has been a great deal of concern that in cats and horses, especially pain-free animals, opioids cause excitement (Muir et al., 1978), including box-walking in horses. This stems from pharmacological studies which used much higher doses of opioids than would be used clinically. Sensible clinical doses of opioids can be used quite safely in these species, as excitement is particularly unlikely to occur when pain is present. Dysphoria may occur after higher doses of opioids in all species. Use of sedatives such as acepromazine (ACP) or the α2-agonists may help to minimize the excitatory or dysphoric effects of opioids.
Vomiting is most likely to occur after the use of morphine in dogs and cats (KuKanich et al., 2005) and is less likely after administration of other opioids. Vomiting is thought to be due to stimulation of the chemoreceptor trigger zone. Administration of morphine as a premedicant also significantly increases the risk of gastro-oesophageal reflux during general anaesthesia in dogs (Wilson et al., 2005). Administration of ACP 15 minutes before the opioid significantly reduced the incidence of vomiting in dogs (Valverde et al., 2004a). Opioids reduce gastrointestinal motility in most species causing constipation and reduced faecal output (Boscan et al., 2006a). These GI alterations may predispose horses to colic due to pelvic flexure and caecal impactions. Some studies have indicated that perianaesthetic morphine does not increase the incidence of colic in horses (Mircica et al., 2003; Andersen et al., 2006), whereas others have reported an increased risk (Senior et al., 2004, 2006). However, other drugs, such as the α2-agonists and general anaesthetic agents, as well as pain itself, will also cause significant reductions in GI motility.
In general, opioids do not have profound cardiovascular effects. Bradycardia may occur, particularly after high doses of the full µ agonists, thought to be due to activation of the parasympathetic nervous system and can thus usually be treated successfully with anticholinergics. There may be a direct negative chronotropic effect of some potent full µ agonists such as remifentanil in humans which is resistant to anticholinergic administration (Tirel et al., 2005), but otherwise there are minimal direct cardiac effects attributed to opioid administration. In horses, hypertension often occurs after opioids (Muir et al., 1978).
Hyperthermia (up to 40°C) after use of opioids has been reported in cats (Posner et al., 2007, 2010). Mydriasis occurs in cats, whereas opioids cause miosis in dogs.
Pruritus has been reported after epidural opioids (especially morphine) and is a major problem in humans. There are occasional reports of epidural morphine-induced pruritus in veterinary species (Burford & Corley, 2006). Urinary retention may also occur after epidural opioids. It is usually self-limiting, but requires careful nursing care and bladder expression until normal function resumes. Delayed hair regrowth over the site of epidural opioid administration is also commonly reported.
Histamine release occurs after IV injection of pethidine (meperidine) and occasionally after rapid IV injection of morphine, resulting in hypotension (Guedes et al., 2006). The other opioids are much less likely to induce histamine release after IV injection.
All opioids cross the placental barrier and may result in respiratory depression and sedation in the neonate if used at parturition. An opioid antagonist such as naloxone may be used to antagonize these effects in the neonate.
Many of the side effects are thought to be mediated by peripheral opioid receptors, raising the possibility of the use of peripheral opioid antagonists which are unable to cross the blood–brain barrier, such as methylnaltrexone, to reduce side effects without affecting the degree of analgesia produced (van Hoogmoed & Boscan, 2005; Boscan et al., 2006b).
Opioid-induced hyperalgesia (OIH) is a recognized phenomenon in humans and rodents associated with both acute and chronic opioid use, resulting in a paradoxical increased sensitivity to pain after opioid administration (Angst & Clark, 2006; Lee et al., 2011). The underlying pathophysiological mechanisms are complex and involve many different pathways and receptors, including interactions between the NMDA receptor, 5-HT receptors, substance P and nitric oxide among others. In humans and rodents, it is thought to have a genetic basis, associated with alterations in genes encoding for peripheral β2-adrenoceptors and drug transporters. It has not been investigated to any great extent in veterinary species.
Drugs available
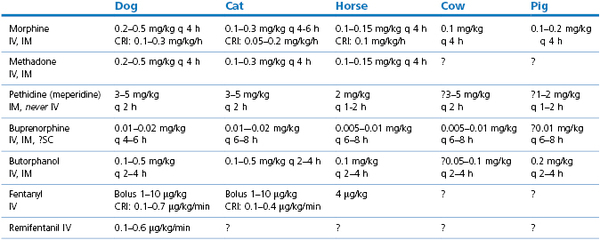
Doses of commonly used opioids are listed in Table 5.2.
Morphine
Morphine is still considered the ‘gold standard’ opioid to which all other drugs are compared. It produces profound analgesia mediated via µ (and possibly κ) agonist activity. It produces euphoria and is addictive in humans and thus falls into Schedule 2 of the UK controlled drugs legislation.
Onset time is around 10–15 minutes after IV administration and up to 30 minutes after IM administration. Morphine is most commonly administered every 4 hours in dogs and horses and every 4–6 hours in cats. Individual variations in duration of action reinforce the need for frequent pain assessment to titrate an analgesic regimen to effect.
The major metabolite in humans is morphine-6-glucuronide which may contribute to morphine’s analgesic efficacy. Morphine-6-glucuronide has not been detected in dogs following morphine administration (KuKanich et al., 2005).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


