Chapter 38 Nonsteroidal Antiinflammatory Analgesics
Nonsteroidal antiinflammatory analgesics (NSAIAs) are a group of pharmaceutical agents that possess both analgesic and antiinflammatory properties. The NSAIAs are frequently used in human and veterinary medicine to relieve mild, moderate and severe pain associated with surgery, inflammatory conditions, and osteoarthritis. The efficacy of many NSAIAs are equal or superior to the pure μ-opioid agonists (e.g., oxymorphone, morphine, hydromorphone, meperidine), and butorphanol or buprenorphine in managing postoperative pain.1–15 When used in combination with opioids, a synergistic effect is achieved and may allow for reduced dosing of the opioid in mild to moderate, but not in severe, pain states where higher opioid dosages may still be necessary. In addition to their centrally acting antinociceptive effect, NSAIAs concentrate in inflamed joints and tissues with a duration of effect of 12 to 24 hours.16 The duration and efficacy of the NSAIAs make them ideal for treating acute1–15 and chronic pain17–28 in veterinary patients; however, because of their potential for harm, patient and NSAIA selection must be considered prior to administration. More detailed reviews of NSAIAs have been published elsewhere.29–39
Pharmacology of Eicosanoid
Cyclooxygenase (COX) enzymes oxidize arachidonic acid to various eicosanoids and related compounds—prostanoids, prostacyclin, thromboxanes, and leukotrienes (Fig. 38-1).40 COX-1 and COX-2 are constitutively expressed throughout the body. Their functions are determined by the specific sites and locations within tissues and cells, and the stimulus triggering their production. Canine and human COX-1 and COX-2 isoenzymes share 96% and 93% DNA sequence homology, respectively. The isoenzyme distribution does differ amongst the species, therefore extrapolation to specific anatomic function may not be appropriate.
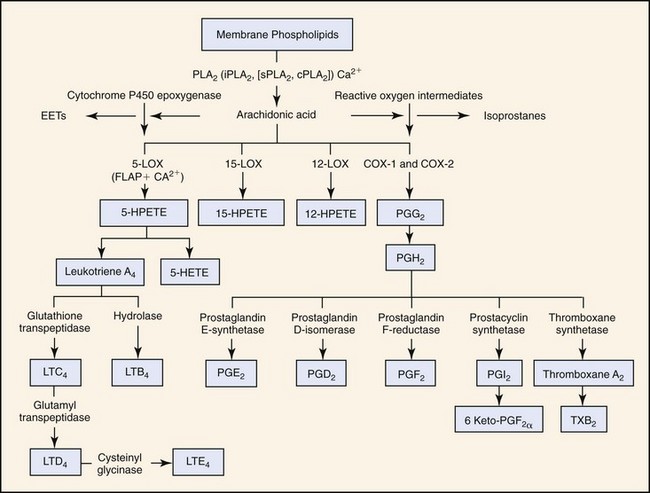
COX-1, present in all tissues, ultimately converts arachidonic acid into prostanoids, thromboxanes, and prostaglandins (PGE2, PGF2, PGI2, and PGD2), which are involved in many homeostatic functions and which are organ specific.46 For example, prostaglandin (PG) activity throughout the body is important in maintaining smooth muscle tone, modulation of vascular tone, and regulation of body temperature46,52; PGE2 maintenance of the gastric mucosal barrier properties; and thromboxane A2 (TXA2), released by platelets in response to vessel injury is responsible for primary plug formation, thrombosis, and primary hemostasis.
COX-2 is involved in many pain and inflammatory states, and as an essential enzyme in many constitutive functions. Prostacyclin (PGI2) is a COX-2 metabolite that, when released from the endothelium, is antithrombotic in modulating TXA2 and maintains a nonthrombotic barrier between the vessel wall and the blood. Prostacyclin is a potent vasodilator that is regulated in several organ systems. In the kidney, for example, this includes the structures that play an essential role in renal blood flow associated with renin activity, fluid–electrolyte homeostasis,31,53,57 and nephron maturation.53,56 COX-2 enzyme also has important constitutive functions in bone metabolism; nerve, brain, ovarian, and uterine function; and intestinal mucosa healing.52,55
COX-3, a COX-1 variant, is expressed in the brain and its microvasculature in the dog.46,49,50 Its importance in humans has not yet been established. The COX-3 isoenzyme is associated with generation of fever and appears to be more sensitive to NSAIAs that are analgesic and antipyretic, but which have low antiinflammatory activity.44
PGs serve as both inflammatory and antiinflammatory mediators where COX-2–derived PG (the PGJ2) functions in resolution of inflammation.47,50,51 PGJ2 interacts with nuclear receptors that comprise the peroxisome proliferator-activating receptor (PPAR) family, particularly PPAR-γ. Activation of PPAR-γ transrepresses the activation of many transcription factors, including nuclear factor κβ (NF-κβ), an important promoter in many inflammatory responses. PPAR-γ is found in macrophages, dendritic cells, and B and T lymphocytes with potential roles in regulating inflammation and immunomodulation.50,51
Leukotrienes produced in the 5-LOX (5-lipoxygenase) cascade are also involved in the inflammatory response. Arachidonic acid is converted in a two-step process into the conjugated triene epoxide leukotriene (LT) A4, the most biologically important form of LT (see Fig. 38-1).47,62–64 LTA4 is subsequently metabolized to LTB4, LTC4, and LTD4. Cells known to express 5-LOX include circulating polymorphonuclear leukocytes (PMNs), monocytes, basophils, eosinophils, tissue macrophages, and mast cells. These cells release LTA4 and participate in the transcellular biosynthesis of either LTC4 or LTB4.65 As with the prostanoids, it is impossible to list all the activities of the LTs as their function is also dependent on organ involvement. With inhibition of COX-1 or COX-2 by NSAIAs, there is the potential of conversion of arachidonic acid into leukotriene B4 via the 5-LOX pathway.39,47,66–68
In addition to the antiinflammatory functions of the PGs, endogenous antiinflammatory mechanisms in the LOX pathway also exist. These consist of small chemical mediators, or autacoids, that play a key role in controlling inflammation by inhibiting PMN recruitment and enhancing monocyte activity.64 Arachidonic acid–derived lipoxins (LXs), particularly LXA4, have been identified as antiinflammatory mediators, illustrating that the LOX pathway also has a dual proinflammatory and antiinflammatory role. These endogenous “antiinflammatory” products are also referred to as resolvins.51 Resolvins, by definition, are endogenously generated within the inflammatory resolution phase and downregulate leukocytic exudate cell numbers.51,82
Cyclooxygenase and the Gastrointestinal Tract
Chapters 45 and 56 describe the mechanisms of gastric and intestinal mucosal protection. The prostaglandin-conferred component is extremely important in the overall protection of the gastrointestinal tract. Many of the effects of the prostaglandins in the gastric mucosa were discovered retrospectively as a result of the side effects associated with NSAIA administration.
Cyclooxygenase-1
The COX-1–derived prostaglandins are responsible for gastric mucosal protection through vasodilation, stimulation, and secretion of gastroduodenal mucus and bicarbonate, and forming a protective barrier to acid injury. These prostaglandins help protect the stomach’s mucosal integrity during periods of vasoconstriction and the daily “trauma” of ingestion, digestion, and emptying. The cytoprotective and trophic functions are important in wound healing secondary to mucosal damage of foods transiting through the stomach. Exogenous administration of prostaglandins have been shown to inhibit gastric acid secretion, stimulate gastroduodenal mucus and bicarbonate secretion, maintain or enhance gastric mucosal perfusion, and have a trophic effect on the gastric mucosa.51a
Cyclooxygenase-2
Some studies also show a protective role for COX-2 in the maintenance of gastrointestinal integrity. For example, upregulation of COX-2 is identified in the duodenum in response to mucosal injury.52,54
Pathophysiology of Nonsteroidal Antiinflammatory Analgesic–Induced Gastritis and Gastric Ulcers
NSAIA gastropathy describes the subepithelial hemorrhages, gastroduodenal erosions, and ulcerations that can occur secondary to NSAIA administration.54a Lesions range from simple erosions affecting only the mucosal layer of the stomach to ulceration affecting the submucosa and deeper layers. There are several studies in dogs reporting gastric ulceration associated with aspirin, flunixin meglumine, phenylbutazone, meclofenamic acid, piroxicam, naproxen and ibuprofen,54b and more recently in association with combination or sequential therapy, or inappropriate dosing of deracoxib73 and meloxicam.74 The inhibition of COX-1 is the principal mechanism responsible for the development of gastric lesions. When a synthetic PGE1 (misoprostol) and an NSAIA (aspirin) with known COX-1 selectivity were administered concurrently, dogs developed significantly fewer gastroduodenal lesions than dogs receiving aspirin alone, but mild gastroduodenal lesions still developed in some of the dogs despite the administration of misoprostol.54c,54d Despite the importance of COX-1, COX-2 inhibition may also contribute to the pathophysiology of gastric ulceration. A study evaluating the effects of various COX inhibitors (a selective COX-1 inhibitor, a selective COX-2 inhibitor, a preferential COX-2 inhibitor, and nonselective inhibitor) showed that prolonged healing and a decrease in gastric blood flow were noted in all treatment groups compared with controls. The effect was most profound in the selective COX-1 and nonselective inhibitor treatment groups. The coadministration of synthetic PGE2 in similar treatment groups facilitated healing. Analysis of mucosal (COX-1) and (COX-2) of the saline-treated group revealed stable levels of COX-1 at all times during the study; but COX-2 levels were not detectable in intact mucosa. In the ulcerated mucosa of the NSAIA-treated rats, COX-2 was present at all times and peaked on day 7 of a 14-day test period.54e This study suggests that COX-2 may play an important role in protection during response to inflammation. In a later study COX-2 was found to be constitutively expressed in canine pyloric and duodenal mucosa. NSAIAs may amplify or decrease the endogenous antiinflammatory response. Aspirin is more COX-1 selective and can impair many components of mucosal defense and enhances leukocyte adherence within the gastric and mesenteric microcirculation.54f However, with chronic use of aspirin there is an adaptation of the gastric mucosa, which occurs at approximately 14 days, and is associated with a marked upregulation of COX-2 expression and lipoxin production. This lipoxin is termed aspirin-triggered lipoxin (ATL). Aspirin is unique among current therapies in that it acetylates the COX-2 enzyme thereby enabling the biosynthesis of 15(R)-hydroxyeicosatetraenoic acid (15[R]-HETE), which is converted to ATL by 5-LOX. Inhibition of either of the COX-2 or 5-LOX enzymes results in blockade of ATL synthesis. LXA4 and ATL (a carbon-15 epimer of LX) attenuates aspirin-induced leukocyte adherence, whereas selective COX-2 inhibitors augment aspirin-induced damage and leukocyte adherence to the endothelium of mesenteric venules in rats.54f
NSAIAs are excreted at varying rates, depending upon the metabolic pathway and extent of enterohepatic circulation. There are many species differences in drug elimination among the NSAIAs. For some drugs, the enterohepatic cycling may increase the risk of toxicosis because of the persistent local effects of the drug on the intestinal mucosa through repeated cycling in the biliary system.34
For NSAIAs that are administered orally, a topical or local effect on the gastric mucosa may also be responsible for the development of erosions/ulcers. In the acidic environment of the stomach, NSAIAs exist in a nonionized form, which are relatively lipophilic and will readily permeate gastric mucosal cells. Here the NSAIA is effectively trapped and can lead to an increase in osmotic pressure causing swelling and cell lysis. The disruption of the gastric mucosal barrier, may allow the diffusion of gastric acid into the submucosa, contributing to further damage.54b The effect of NSAIAs on the microcirculation to the stomach may be another component of the pathogenesis of gastropathy. Aspirin has been shown to cause a focal decrease in mucosal blood flow at the sites of subsequent ulcer formation.54g Some of these alterations in blood flow are likely mediated through the systemic inhibition of PG synthesis, or local effects of NSAIAs. Another potential mechanism affecting the microcirculation is the increase in neutrophil adherence to the vascular endothelium after NSAIA administration resulting in direct obstruction of the microvasculature, or by causing damage to the endothelium and epithelium by releasing proteases and free radicals.54b
Signs of Gastric Ulceration
Clinical signs of gastric ulceration in dogs include vomiting, hematemesis, melena, abdominal pain, pallor of the mucous membranes, lethargy, weakness, collapse and anorexia. Interestingly, experimental studies of NSAIAs reported clinical signs of gastric ulceration to be inconsistently present in dogs with endoscopic or gross evidence of gastrointestinal lesions.54h Some dogs remained bright and alert with no signs of abdominal pain. These findings are consistent with reports in humans where most cases of NSAIA-induced gastric ulceration go undiagnosed until they are life-threatening, complicated ulcers. The delay between the development of gastric ulceration and the observation of clinical signs may be related to unique properties of the NSAIA, or clinical signs may not develop until a deep ulcer erodes into a large blood vessel. Poor correlation between gastrointestinal lesions observed by endoscopy and clinical signs following treatment with carprofen, ketoprofen, or meloxicam have been reported in other studies. These lesions were mild to moderate in severity and did not differ statistically from placebo. The initial signs of vomiting and anorexia are frequently due to gastric irritation or inflammation. Because of the requirement of a severe lesion prior to the presence of hemorrhage or acute kidney injury, it is essential that owners be warned to stop NSAIA administration and seek immediate advice from the family veterinarian should the dog or cat develop vomiting or anorexia.
Role of Prostaglandins and Antiprostaglandins in Nociception
Nonsteroidal antiinflammatory analgesics inhibit COX-1, COX-2, or both, or COX-3 resulting in reduced PG synthesis. COX-2 is inducible and synthesized by macrophages and inflammatory cells, potentially increasing by 20-fold over baseline, especially in injured tissue and inflammatory conditions such as osteoarthritis.42 Increased COX expression stimulates prostanoid production and these compounds serve as mediators of inflammation and amplifiers of nociceptive input in the peripheral and central nervous systems.42 By this mechanism, COX-2 is responsible for a significant amount of pain and hyperalgesia following tissue injury. COX-1 is increased approximately two- or threefold during tissue injury, and may also generate PGs at sites of inflammation (e.g., joints, skin, gastrointestinal tract). COX-1 is present within the central nervous system and active in pain transmission. In addition to the peripheral effect, a significant part of the NSAIA antinociceptive effect is exerted at the spinal cord and supraspinal levels.41–47 This action, in addition to pain relief, may account for the observed overall well-being and improved appetite of patients receiving injectable NSAIAs for relief of acute pain.
Pain management should be considered in its physiologic context as pain pathways involve COX-1 or COX-2 genes predominantly, both of which are differentially expressed. .COX-1 selective NSAIAs, for example, are superior to COX-2 selective NSAIAs at inhibiting visceronociception and visceronociception is also greatly reduced in COX-1 but not COX-2 knockout mice. Visceral pain may be mediated by intraperitoneal receptors on sensory fibers by COX-1–produced prostacyclin. These studies concluded that peripheral COX-1 mediates nociception in slowly developing pain in mice, such as in visceral pain, and central COX-1 may be involved in rapidly transmitted, nonvisceral pain, such as that caused by thermal stimulation. Interestingly, there may be gender differences, as in Ballou’s mouse model, COX-2 was also found to mediate visceral nociception, but only in female mice.47a The analgesic potency of a range of NSAIAs in relieving tooth-extraction pain in humans correlates closely with increasing selectivity toward COX-1 rather than COX-2. These findings highlight the importance of both COX-1 and COX-2 contributing to pain and the selectivity of NSAIAs in treating painful conditions. The veterinary COX-2 preferential (COX-2 inhibition with some COX-1 sparing) NSAIAs may have similar activity.
Early research emphasis was placed on development of medications that inhibit COX-2 activity and spare constitutive COX-1 function. Theoretically, COX-2–selective NSAIAs should be effective, with potentially fewer adverse effects in the management of pain. Unfortunately, this complex biologic system is still not clearly defined and current knowledge indicates a role for both in nociceptive transmission and constitutive functions,47,48 and the notion of “good versus bad COX” is probably too simplistic.34 In addition to the COX-2 role in inflammation, aberrantly upregulated COX-2 expression is increasingly implicated in the pathogenesis of a number of epithelial cell carcinomas, including those of the colon and esophagus.58,59 COX-2 inhibitors are being studied as potential anticarcinogenic agents. Although glucocorticoids are not analgesics, the COX-2 gene is glucocorticoid sensitive, in that it is reduced following administration of glucocorticoids, which may partially explain the antiinflammatory and analgesic effects of this class of medications.47,58,60
The COX-3 isoenzyme has been proposed as a target of the analgesic/antipyretic agents acetaminophen and dipyrone.46,49,50 Both acetaminophen and dipyrone have minimal effect on COX-1 and COX-2,46 and are frequently used to reduce fever in animals with little gastrointestinal or renal adverse effects. The COX-3 isoenzyme is more sensitive to NSAIAs that are analgesic and antipyretic, but which have low antiinflammatory activity. This fact emphasizes the different niche for NSAIA therapy in managing pain of differing etiology. As the COX-3 isoenzyme is derived from the COX-1 gene, this suggests that the COX-1 gene plays an integral role in pain and/or fever depending on the physiologic context.44
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


