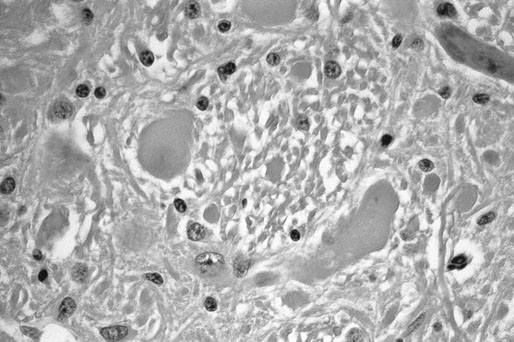
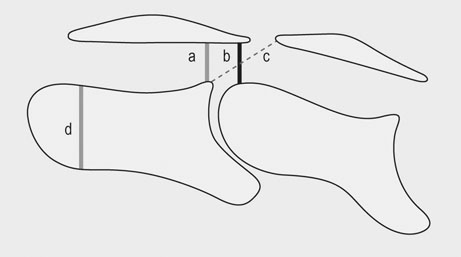
There are two recognized forms of the disease. Type 1 CVM results in dynamic compression with flexion of the neck and most often occurs in the mid-cervical region (C3-C5). Type I CVM most commonly occurs in young, rapidly growing, male horses. Bony abnormalities are similar to those seen in osteochondrosis and epiphysitis of the appendicular skeleton with cartilage retention and dissecting lesions of the joint surface.1,2 Type 2 CVM is a static stenosis of the canal most commonly occurring in the caudal neck (C5-T1). Type 2 is reported to occur more commonly in older horses though young horses are also affected. Causes of static compression include cranioventral extension of the dorsal lamina, caudodorsal extension of the caudal vertebral body, hypertrophy of the ligamentum flavum, enlargement of the articular processes of the cervical facets and, less commonly, disc herniation and synovial cyst formation intervertebral disk prolapsed and diskospondylitis in a horse.3–5 The underlying etiology of CVM is incompletely understood and is likely multifactorial. Genetics, trauma, high energy diets and micromineral imbalances are all suspect to play a role in the pathogenesis. In addition, similar to humans and other species it is likely that progressive age related degeneration, repetitive movement, instability and trauma may also be factors in the development of this syndrome. Cervical vertebral malformation is most commonly seen in the young, large, rapidly growing, male phenotype with Thoroughbred and Warmblood breeds over-represented.6,7 While genetics must certainly be a factor in the development of CVM a study breeding CVM-affected horses to CVM-affected horses resulted in an increased incidence of osteochondrosis of the appendicular skeleton without an increased incidence of CVM supporting a similar pathogenesis in these two syndromes.8 In addition, a study of affected and unaffected Thoroughbreds in Great Britain failed to identify a genetic mode of inheritance.9 It is likely that both genetic and environmental factors including diet play a role in the development of CVM. Foals identified as having risk factors for CVM (radiographic changes and or neurologic signs) showed improvement in both radiographic changes and neurologic function when fed a restricted diet and confined to limit exercise. The restricted diet was low in protein and energy with vitamin and mineral composition adjusted for the age and weight of the individual foal.10 An association between both CVM and osteochondrosis and mineral balance has also been reported.11,12 Unfortunately because many affected horses are not identified till later in life it is unlikely that dietary modification at that time will affect growth and outcome. Four commonly recognized bony abnormalities may be seen on radiographs including flare of the caudal vertebral epiphysis of the vertebral body, extension of the caudal edge of the dorsal arch of the vertebral body, osteoarthritis of the articular processes and malalignment of adjacent vertebrae (Fig. 24.1).13 Because these changes can be seen in unaffected horses these findings are supportive but not definitive for a diagnosis of CVM. In addition to evaluating radiographs for bony changes, narrowing of the canal can be assessed by measuring intra- and inter-vertebral minimal sagittal diameter ratios (MSDR).13–15 Minimal sagittal diameter ratios compare the maximum width of the canal within the vertebrae (Intra-) or between vertebrae (inter-) to the maximum width of the body of the cranial cervical vertebrae allowing the assessment of the width of the canal while controlling for magnification effects (Fig. 24.2). It is critical that a true lateral be taken to ensure accuracy. An abnormally low ratio is associated with narrowing of the vertebral canal and suspicious for compression of the spinal cord in this region. Multiple studies have attempted to predict the likelihood of CVM in suspect cases using scoring systems that utilize bony abnormalities alone or in combination with intra- and inter-MSDRs.13–18 Papageorges et al concluded that lateral radiographs was not useful in evaluating horses for CVM. In this study only bony abnormalities were scored without determining minimal sagittal diameter ratios. The finding that bony changes alone are not diagnostic for CVM is supported by a recent study determining that cervical facets at C5-C6 and C6-C7 enlarge with age; this enlargement did not correlate with neurologic deficits.19 Mayhew et al reported on a radiographic scoring system in foals and weanlings that utilized both bony abnormalities and MSDRs. In this study a high score (increased number of abnormalities) was associated with CVM. The authors used the results of this study to determine which individuals were placed on a restricted diet and reported a decrease in both the radiographic scores and neurologic symptoms.10 The authors noted that certain criteria (angulation and stenosis based on MSDRs) were more accurate in predicting CVM than other criteria. Rush-Moore et al evaluated the accuracy of an intra-vertebral minimal sagittal diameter in predicting CVM in 50 affected and 50 control horse. To be included affected horses had to have histopathologic evidence of CVM at post mortem examination. They concluded that a MSDR cutoff of 0.52 for C4, C5 and C6 and 0.56 for C7 resulted in the highest sensitivity and specificity. Interestingly, while an abnormal MSDR showed strong correlation in predicting a CVM-affected individual, it was less accurate in predicting the specific site of compression leaving the authors to recommend myelography when planning surgical stabilization. A later study further confirmed the utility of intra- and inter-vertebral minimal sagittal diameters for predicting CVM using a cutoff value of 0.485.15 Because there is some overlap in MSDR values in affected and unaffected sites a myelogram is strongly recommended to confirm both disease and specific location of compression. To further confuse the interpretation of MSDRs, a recent report showed significant inter and intra-observer variability in measurement of cervical vertebral ratios putting the accuracy of these measurements into question.18 Given these findings, it is reasonable to conclude that lateral radiographic assessment including measurement of minimum sagittal diameter ratios is useful as a screening tool, but should not be used as the sole diagnostic criteria when evaluating a horse with suspect cervical vertebral malformation. The technique for performing a myelogram has been well described.20 Briefly, the horse is placed in lateral recumbency and the poll area clipped and prepped. The land marks for the site are the cranial border of the wings of the atlas on dorsal midline. The poll is flexed and a 3.5 inch 18 gauge needles with a stylet is advanced angled towards the lower incisors. The dura mater must be penetrated and may produce a popping sound when entered. Cerebral spinal fluid will drip from the needle once the trochar is removed. Samples of CSF for additional testing should be collected prior to injection of contrast material. The bevel of the needle is faced caudally and ~40–50 mls of Iohexol injected slowly over a five minute period. The head and neck should be elevated on a wedge platform for five minutes to facilitate flow of contrast down the spinal canal. Radiographic images should be taken in neutral and flexed positions; some clinicians will also take images of the lower spine (C6-T1) with extension though this may not be necessary (Fig. 24.3). While extreme flexion and extension may be necessary to achieve a diagnosis it may result in exacerbate of clinical signs and should be performed with caution and only if neutral views are non-diagnostic. Complications include depression, deterioration of neurologic condition, fever, seizures, corneal ulcers, muscle fasciculations, hyperesthesia, stiff neck and meningitis.20–24 Complications appear to occur less frequently with the use of Iohexol. Multiple diagnostic criteria have been published. The most common criteria is the finding of a 50% or more height reduction of the minimal dorsal myelographic column (DMC) at the intervertebral site compared to the maximal diameter at the level of the cranial vertebral body. Other reported criteria include the empiric <2 mm DMC and the dural diameter (DD) reduction which is measured by comparing the maximum DD at the cranial vertebral body to the minimum DD at the intervertebral joint. The data behind the development of these criteria is unclear and with one exception, they have not been validated.25 A recent report suggests that diagnostic criteria for compression should vary with the vertebral site and neck position (neutral or flexed).26 The authors conclude that a single diagnostic criteria is not adequate when analyzing a myelographic study. The 50% reduction in the DMC is adequate when evaluating the neutral views of the mid-cervical vertebrae (C3-C6) as it has a high specificity (low false-positive rate) but that a more conservative (greater reduction of the DMC) should be used in the flexed view to avoid an increase in false-positive diagnosis. The authors suggest that a cut-off of 50% can be used for a flexed view of C6-C7 with a more conservative cut-off (70%) used for this site in a neutral position. Alternatively, a 25% reduction in the DD may be used with either neutral or flexed views at C6-C7 with an acceptable sensitivity and specificity. Medical treatment consists primarily of stall rest and anti-inflammatories including NSAIDs and DMSO. Glucocorticoids may be beneficial if an acute exacerbation or injury occurs. Given the underlying pathology medical therapy may temporarily improve clinical signs but is unlikely to resolve the disease. Injection of corticosteroids into the cervical facets may improve clinical signs especially if evidence of neck pain or spinal root compression exists. No data is available on the long term outcome with medical therapy alone. In a multicenter retrospective of long term outcome the majority of severely-affected horses were euthanized or had surgical stabilization; those mildly affected were more likely to be discharged without surgery.27 Surgical stabilization or decompression is the most effective treatment for CVM.28–32 Ventral surgical stabilization is recommended for Type 1 CVM when compression is the result of dynamic instability of the intervertebral joint. A stainless steel implant and bone graft are used to fuse the ventral vertebral bodies at the affected joint. Dorsal laminectomy is recommended to immediately decompress the spinal cord when static stenosis is the cause of the neurologic deficits. Removal of the dorsal lamina, ligamentum flavum and joint casule can result in immediate relief. Ventral stabilization can also be beneficial in cases of static stenosis as fusion may result in bony remodeling and atrophy of soft tissue with delayed decompression of the site. Surgical treatment is ideal for animals with acute disease as permanent deficits are more likely with chronic compression. In a retrospective report evaluating surgical treatment of 73 cases using ventral stabilization or dorsal laminectomy neurologic function improved in 77% of horses with 46% returning to athletic performance.28 Horses with a shorter duration of signs (<1 month) and only one affected site had a better prognosis than those with longer duration and multiple sites of compression. Other studies report similar results with improvement in neurologic function from 44 to 90% and a better prognosis when only one site is affected.29–32 The risk of surgical complication is relatively high with approximately 10% of horses not surviving to discharge. The successful stabilization of Type 2 CVM using a ventral locking compression plate in a three-month-old foal has also been reported.33 Equine protozoal myelitis, originally termed segmental myelitis, was first recognized as a cause of neurologic disease in the 1960’s.34 The agent, Sarcocystis neurona, was subsequently cultured from the spinal cord of an affected horse. More recently the protozoal agent Neospora hughesi has been identified in CNS tissue from neurologically affected horses; however it appears to be an infrequent cause of EPM.35 Equine protozoal myelitis is recognized in the Western Hemisphere its range corresponding to the range of various opossum species. The opossum is the definitive host of S. neurona and becomes infected after eating muscle containing sarcocysts.36,37 Sporocysts are passed in the feces of the definitive host and ingested by the intermediate hosts including cats, skunks, armadillos and raccoons.38–40 Currently, the raccoon is thought to be the most significant intermediate host in North America. After ingestion sporozoits are released and penetrate the gut wall of the intermediate host entering the vascular endothelial cells. Asexual reproduction in the endothelial cell results in release of merozoites into the blood stream. In the natural host, merozoites eventually enter skeletal muscle forming sarcocysts. The life cycle is completed when the opossum eats the dead muscle of the infected intermediate host. Horses are thought to become infected through ingestion of opossum feces containing sporocysts contaminating their feed or environment. The exact route of migration from ingestion to infection of the CNS is unknown but parasitemia has been detected in immunocompromised and one immunocompetent horse.41,42 The horse is considered a dead end host though mature sarcocysts have been found in the tongue muscle of one horse challenging this assumption.43 The pathogenesis of S. neurona in equids is incompletely understood as, despite an apparent high rate of exposure, clinical illness is much less common.44–48 In a USDA study the average incidence of EPM was 14 cases per 10 000 horses per year.49 Horses used for competition or racing had a much higher incidence than those used for pleasure or breeding. Certain breeds including Thoroughbreds, Quarter Horses and Standardbreds are over-represented as are young horses, though all breeds and ages can be affected.50,51 Parasite dose and strain, immune function and physiologic stress (including strenuous exercise) are all thought to play a role in pathogenesis. A common model for inducing EPM is long-distance transport further supporting the role of stress in the pathogenesis of this disease.52 The Western Blot (WB) was the first test to be developed and can be performed on both serum and CSF.53 Because of the high rate of exposure without clinical disease serum testing is useful as a screening test as seronegative horses are unlikely to have EPM. In one study the sensitivity and specificity of the WB was found to be the strongest in neurologic horses.54 False positive CSF titers may occur due to passive movement of antibody across the BBB, due to iatrogenic blood contamination or due to subclinical infection. A modified Western blot (mWB) was developed to try to decrease false positive results due to cross reaction with other Sarcocystis antigens. Sensitivity and specificity of this mWB was reported to be 100% and 98% by the test developers and 89 and 69% in another study.55,56 An additional modified Western blot is available that reports the intensity of the staining result as a relative quotient. This modification has not been shown to improve diagnostic value. More recently an enzyme linked immunosorbent assay (ELISA) was developed against the snSAG-1 protein of S. neurona.57 An independent study determined the sensitivity and specificity of this test to be poor.58 In addition, not all S. neurona isolates produce the SAG-1 protein. A separate research group developed and evaluated a series of ELISA tests against multiple S. neurona surface antigens (sn SAG1-4). The ELISA against the snSAG-2 protein had the highest sensitivity and specificity, 95.5 and 92.9% respectively, using serum and CSF from cases of histopathologically confirmed EPM.59 A whole organism S.neurona immunofluorescent antibody test (IFAT) is also currently available. In a study comparing the IFAT to the WB and the mWB, the IFAT was reported to have a higher specificity than either WB test.56,60 Iatrogenic blood contamination of CSF during collection has been shown to confound results with as little as 11 RBC per microliter causing a false positive CSF Western blot.61 Evaluation of both serum and CSF as well as performing CSF cytology are recommended when running immunodiagnostic testing for EPM. In addition, because proteins, including antibody, can passively cross the BBB, antibody levels in the CSF can occur in a healthy individual. In theory this passive movement results in CSF antibody levels in proportion to those in the blood, whereas true CNS infection should result in a much higher proportion of specific antibody in the CSF relative to that seen with passive movement from the blood. Consequently, recommendations for interpretation of immunodiagnostic testing of the CSF are to include CSF indices to distinguish intrathecal production from passively acquired antibody in the CSF.62 Because there is no ideal diagnostic test for EPM, it is important to establish an accurate list of differentials by considering the signalment, history and progression of neurologic signs, time of year, vaccination history, location and use of the affected animal. Additional diagnostics may include cervical films to rule out cervical vertebral malformation, PCR on CSF to rule out EHV-1 and PCR and/or immunodiagnostic testing of serum and CSF to rule out other infectious diseases such as West Nile Virus or Equine encephalitidies. A diagnosis of EPM is best approached using clinical exam and findings of neurologic signs that are compatible with EPM, ruling out other potential causes of the neurologic symptoms and finally a positive immunodiagnostic test on the CSF.63 Because it is difficult to experimentally reproduce clinical disease in the horse, efficacy of current therapeutics is based on in vitro assays, experimental infections in laboratory animals and clinical trials. There are currently three classes of medications recommended for the treatment of EPM. The first recommended treatment for EPM was a combination of sulfonamides (20 mg/kg) (with or without trimethoprim) and pyrimethamine (1mg/kg).64 This combination is synergistic blocking protozoal folate synthesis at multiple steps (Beech 1974). In a multicenter field study of clinical cases treated with a combination of pyrimethamine and sulfadiazine (Rebalance, Animal Health Pharmaceuticals, LLC), an overall success rate (defined as an improvement of 1 grade in neurologic deficits or conversion to a negative CSF Western blot) of 53% was reported.65 Duration of treatment was a minimum of 90 days. Reported side effects in horses receiving the label dose included anemia (22%), leucopenia (19%), neutropenia (5%) and thrombocytopenia (3%). Bone marrow suppression is thought to be due to folate deficiency. In addition, congenital defects in foals born to mares treated with sulfonamides and pyrimethamine during pregnancy have been reported.66 Horses treated with this combination should be fed good quality green forage (high in folate) to decrease the risk of side effects. Folate supplementation is not thought to be beneficial and may increase the risk of toxicity.67,68 Though less expensive than other treatment regimes the duration of treatment and risk of side effects may offset the decreased daily cost. The triazine anticoccidial agent, Ponazuril (Marquis, Bayer Animal Health), was the first FDA approved drug for treatment of EPM. Triazine anticoccidial medications are thought to inhibit the respiratory chain enzymes of the protozoal organism.69 Recommended oral dosing of Ponazuril is at 5 mg/kg once daily for 28 days. Feeding of two ounces of corn oil immediately prior to dosing has been shown to increase the blood levels by 25%. If neurologic deficits persist, many clinicians will recommend a second 30-day course of therapy. The overall success rate of Ponazuril in the treatment of EPM has been reported to be 62%.70 Recently another triazine anticoccidial, Diclazuril, has received FDA approval (Protazil, Schering-Plough Animal Health). Currently Diclazuril is marketed as a pelleted oral medication though many clinicians report successful treatment of suspect EPM cases using a compounded intravenous form of Diclazuril. In a multicenter study of clinical cases treated with Protazil the overall success rate was 67% though the percentage dropped to less than half when reviewed by additional experts blinded to the timing of the video exam of enrolled cases.71 The thiazolide derivative, Nitazoxanide (Navigator, IDEXX Pharmaceuticals) was approved in 2003 and marketed as the only medication effective in killing S. neurona. Adverse reactions including diarrhea, colic, edema and laminitis were common with this medication leading to recommendations to give one half the recommended dose for five days before starting the full dosing schedule.72,73 Adverse reactions are common due, in part to Nitazoxanide disturbance of the gastrointestinal flora. Currently Nitazoxanide is no longer on the market. In vitro efficacy of additional thiazolide and thiadiazolide derivatives against S. neurona has been reported.74 Whether theses derivatives will cause less adverse reactions remains to be determined. A vaccine was developed against EPM in the mid 1990s. Vaccinated horses may become transiently positive on CSF Western blot making knowledge of vaccination history important when evaluating a suspect case.75 Efficacy remained unproven and the vaccine is no longer commercially available. Pretreatment with ponazuril at 5 mg/kg once daily decreased the incidence of clinical EPM in a challenge model.76 Weekly treatment with 20 mg/kg was also effective in decreasing CSF antibody development in challenged horses. Given the incidence of EPM it is unlikely that prophylactic therapy would be cost effective except in specific circumstances. Current prevention recommendations are primarily targeted on preventing contamination of feeds and environment by S. neurona sporocysts through limiting opossum access to the barn yard and feedstuffs. This can be done by trapping, keeping dogs to discourage opossums from entering the premises and by limiting access to food sources. In particular, keeping equine feedstuffs secure to prevent contamination is strongly recommended. Neuroaxonal dystrophies are a group of degenerative axonopathies recognized in a wide variety of species.77 Degenerative changes are primarily confined to the central nervous system and may be localized to specific nuclei and tracts or diffuse as seen with Equine Degenerative Myeloencephalopathy (EDM) a severe, diffuse form of neuroaxonal dystophy. Both familial and environmental factors are believed to play a role in the pathophysiology of EDM. Equine degenerative myeloencephalopathy has been recognized in most breeds of horses with reports of familial disease in Appaloosas,78 Morgans,79 Standardbreds,80 Mongolian wild horses,81 and Quarter Horses.82,83 Vitamin E deficiency during the rapidly growing stages of life appears to play a role in the development of neuroaxonal dystrophy and EDM particularly in individuals with a familial tendency.78,80,83,84 In a retrospective case/control study risk factors for EDM include housing on dirt lots, exposure of young foals to insecticides and wood preservatives. Housing in green pasture was found to be protective.85 Dietary and serum levels of vitamin E are not always abnormal in affected horses. Serum vitamin E and selenium levels were similar in a retrospective study comparing unaffected to affected horses.84 In humans, ND has been reported as a result of malabsorption of alpha tocopherol from the gastrointestinal tract.86 A lack of hepatic vitamin E transfer protein has also been suggested.87 A deficiency in a metabolic pathway or function of vitamin E cannot be ruled out as some affected individuals appear to have adequate dietary intake of vitamin E. In a study of affected Appaloosa horses, dietary vitamin E was adequate despite low blood vitamin E levels in affected animals.78 A single serum vitamin E level may not be abnormal as levels can vary in affected animals depending on diet and other unknown factors. In addition, serum vitamin E measurement may not directly correlated with total body levels as <1% is found in the blood.88 Consequently, serum vitamin E levels are most useful as a screening test for inadequate dietary intake and not definitive evidence of EDM. Neuraxonal dystrophy was recently reported in a herd of Quarter Horses. Clinical signs of ataxia, dysmetria, a wide based stance with proprioceptive deficits, an incomplete or sluggish menace and a quiet or obtunded demeanor were reported.83 Serum and liver vitamin E levels were low in affected animals. Clinical signs did not appear to progress once recognized. Supplementation of vitamin E the following year appeared to decrease the incidence and severity of new cases. Neurodegenerative changes consistent with neuroaxonal dystrophy were found in multiple nuclei of the brain stem and spinal cord as well as axonal loss, demyelinization and astrogliosis in the long tracts of the spinal cord. A common ancestor was present in most affected horses. Histopathologic examination of the CNS is necessary for a definitive diagnosis of EDM or neuroaxonal dystrophy of Quarter Horses. Histopathologic abnormalities include neuroaxonal degeneration with spheroid formation within axons, pigment deposition, demyelination and astrogliosis (Fig. 24.4). Histopathologic abnormalities may be localized to specific nuclei or diffuse.83,89,90 Vitamin E supplementation is the cornerstone of treatment of EDM and NAD. Unfortunately, supplementation is unlikely to result in significant improvement of affected animals though reports of improvement in individual horses exist. The vast majority of cases appear to stabilize with or without vitamin E supplementation. Vitamin E supplementation has been shown to decrease the number and severity of new cases on breeding farms.78–80,89 Prevention is the key to controlling neuroaxonal dystrophies. Preventative measures should be focused on maintaining adequate vitamin E levels in the diet particularly in breeding stock and young horses. Green pasture and fresh, well cured hays are excellent sources of vitamin E. Yearly analysis of hay and concentrates is recommended to determine vitamin E levels and to assess the need for supplementation. In cases with a history of a familial predisposition it is recommended to outcross or avoid breeding suspect horses. Equine motor neuron disease (EMND) is a diffuse neurodegenerative disorder affecting the somatic, lower motor neurons of horses.91 The disease occurs sporadically with one or two horses affected on a premise though a report of a large numbers of horses affected in one stable exist.92 The Quarter Horse breed is over represented and the risk of EMND increases with age with a peak at 16 years followed by a decline.93 Equine motor neuron disease was first recognized in the Northeastern United States and most cases have been clustered in this region.94 EMND has been reported across the United States, Canada, Europe, the British Isles, Japan and Brazil.92,95–98 The exact etiology of EMND is unknown, though a deficiency in dietary vitamin E is thought to play a role. Epidemiologic studies have identified breed, region of residence, housing on dirt paddocks or stabling without access to green pasture, time living on a premises, feeding of pelleted diets and aged, grass hay, and a lack of supplementation for vitamin E and selenium as risk factors.99,100 Diet and housing alone are not sufficient to cause EMND as many unaffected stable mates are housed and fed similarly to affected individuals. One report of EMND occurring in horses kept on pasture has been published.101 In a case-control study, controlled for age and breed, plasma vitamin E levels were negatively associated with development of EMND.93 This association was strengthened by a later case-control report that controlled for season of diagnosis, age, breed and dietary practices. The authors concluded that while plasma vitamin E levels were confounded by age, breed, type of turnout, and type of concentrate fed, plasma vitamin E levels were still negatively associated with EMND when these factors were controlled.102 Vitamin E is the predominant lipid-soluble antioxidant in mammalian cells. Deficiency of this antioxidant would result in a decreased ability to reduce reactive oxygen species and would make an individual more susceptible to oxidative damage. It is likely that low plasma vitamin E predisposes individuals to EMND but is not the sole cause of this syndrome. Clinical signs are due to degeneration of somatic motor neurons. Histopathologic findings are localized to nuclei of the brain stem and vental horn of the spinal cord and include chromatolysis, swelling, neurofilamentous accumulations, axonal spheroids and eosinophilic inclusions.103,104 Secondary lesions resulting from the loss of motor neurons include peripheral nerve degeneration and severe neurogenic muscle atrophy. Muscles units containing a greater percentage of Type 1 fibers are usually more severely affected.105 These muscle fibers have a higher oxidative activity supporting the conclusion that oxidative stress plays a role in the disease. A pigment retinopathy has also been reported in the majority of cases examined.106 Lesions included a reticulated pigment pattern in the tapetal-nontapetal junction extending throughout the fundus in severe cases. Ceroid lipofuscin was deposited in the retinal pigment epithelia. Visual deficits were not reported in affected animals. Retinal photoreceptor membranes contain a large percentage of polyunsaturated fats making them highly sensitive to oxidative damage; pigment accumulation is typical of that seen with lipid peroxidation. These changes are similar to those produced when rats are fed diets lacking antioxidants such as vitamin E.107 Lipopigment is also seen in the capillary endothelium of the spinal cord in affected horses.99 Clinical signs of acute EMND are muscle fasciculatins, almost constant shifting of weight and increased time in recumbency. Affected animals exhibit weight loss and severe muscle atrophy particularly of the appendicular, antigravity muscles including the triceps, gluteals and quadriceps (Fig. 24.5
Neurologic causes of gait abnormalities in the athletic horse
Cervical vertebral malformation
Epidemiology and pathogenesis
Recognition
Radiographic evaluation
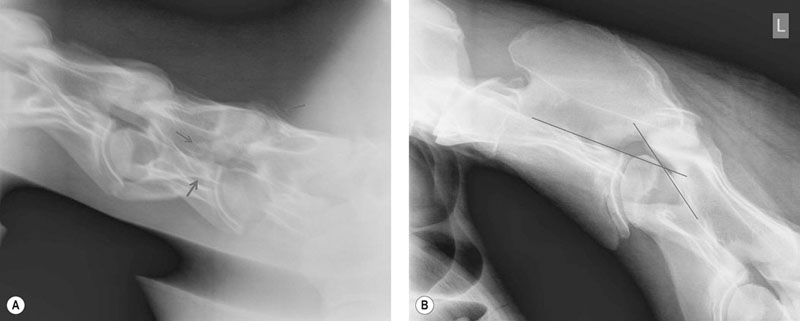
Myelogram
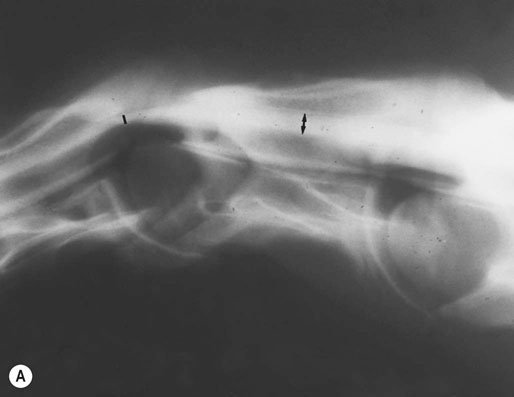
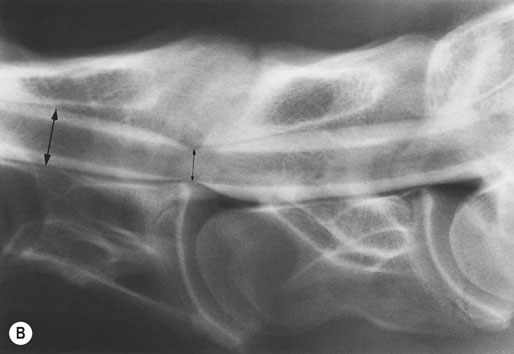
(B) Myelographic examination of C6–C7 with the cervical spine in neutral position. Static spinal cord compression is demonstrated by obliteration of the dorsal and ventral contrast columns. Percent reduction in dural diameter is determined by comparing the intervertebral space (small black arrow) to the midvertebral site cranial to the intervertebral space (large black arrow). Twenty percent or greater is diagnostic of compression (40% reduction in this case).
Treatment and prognosis
Medical treatment
Surgical treatment
Equine protozoal myeloencephalitis
Epidemiology and pathogenesis
Recognition
Diagnosis
Treatment, prevention and prognosis
Treatment
Prevention
Neuroaxonal dystrophy and equine degenerative myeloencephalopathy
Epidemiology and pathophysiology
Recognition
Clinical signs
Diagnosis
Treatment, prevention and prognosis
Equine motor neuron disease
Epidemiology and pathogenesis
Recognition
Clinical signs
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree