Chapter 2 Neuroendocrine control of cardiovascular function
Physiology and pharmacology
NEURONAL REGULATION OF THE CARDIOVASCULAR SYSTEM
Central nervous control
A full discussion of the central nervous system (CNS) control of the cardiovascular system is beyond the scope of this chapter,1 and the following is a simplified account. Baroreceptors (stretch receptors) which detect high pressure within the vascular system are present in the walls of conductance vessels (aortic arch and carotid sinus) and those which determine low pressure (central volume receptors) are in atrial tissue (primarily at its junction with the great veins), pulmonary arteries and ventricles. Afferent inputs to the CNS are transmitted via the glossopharyngeal and vagal nerves which terminate in the nucleus tractus solitarius (NTS). The NTS also receives much information from other areas of the CNS (hypothalamus, cerebellum and cortex) involved to some extent in cardiorespiratory control. Integration of this information occurs in the NTS before it is relayed to the efferent cardiovascular control areas in the medulla of the brain stem (such as the nucleus ambiguus, rostral ventrolateral medulla) which initiate adjustments in vascular tone, heart rate and force of cardiac muscle contraction, mediated via the sympathetic and parasympathetic nervous systems, in order to normalize arterial blood and vascular filling pressures and make them appropriate for the physiological state of the animal.
Drugs which influence CNS control of the cardiovascular system
A number of drugs which are used clinically will affect the activity of centres within the brain controlling the cardiovascular system. Alpha2-adrenoceptor agonists, for example, increase vagal tone to the heart and reduce sympathetic tone to the blood vessels, in part by their effects within the regulatory areas of the brain.2 These actions give rise to the undesirable side effects of bradycardia and hypotension (following transient hypertension) when these drugs are used as analgesic sedatives.
Cardiac glycosides (e.g. digoxin) also have effects on the afferent nerves and the higher centres controlling the cardiovascular system, in addition to their direct effects on the heart and vasculature. Their effects can be very complex, depending on the physiological state of the animal treated, but generally include an increase in parasympathetic tone to the heart and increased baroreceptor stimulation, which produces reflex reduction in sympathetic vasoconstrictor nerve activity.3 Individual variability in the response to cardiac glycosides could well reflect the degree of activation of the putative natural hormone “endogenous digitalis-like substance” which binds to the same receptors.4
Control of vascular tone
Vasomotor nerves
The categories of vasomotor nerves currently recognized are presented in Table 2.1. Efferent nervous control of the vascular system is primarily by the sympathetic vasoconstrictor nerves which tonically increase vascular smooth muscle tone throughout the circulatory system. The postganglionic noradrenergic neurones are distributed to all tissues innervating both arterioles and venules and increasing vascular tone by the action of noradrenaline on α1-adrenoceptors. The density of sympathetic innervation and of α1-adrenoceptors varies with different vascular beds. There is also clear evidence that sympathetic nerves supplying different vascular beds can be preferentially or even exclusively controlled by neurones in the medulla, according to the integrated afferent input.5 Other vasomotor nerves are not tonically active or distributed in such a widespread fashion, but are stimulated under appropriate circumstances (see Table 2.1).6
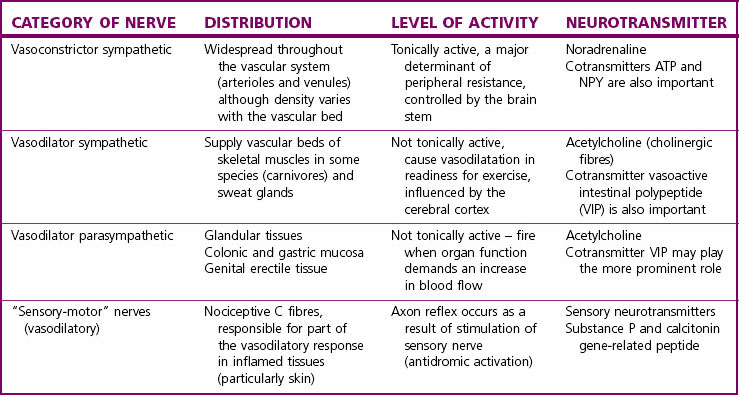
Co-transmission in perivascular nerves
Sympathetic vasoconstrictor nerves also release adenosine 5’ triphosphate (ATP), which is packaged in both large and small dense core vesicles with noradrenaline, and neuropeptide Y (NPY). NPY is preferentially localized in large vesicles.7
In some blood vessels, part or all of the vasoconstrictor response to perivascular nerve stimulation can be shown to be due to ATP, which acts on vascular smooth muscle receptors, distinct from α1-adrenoceptors, namely P2x-purinoceptors. A prazosin-resistant component of the response of equine digital artery to perivascular nerve stimulation has been reported,8 which shows characteristics of a purinergic response. The physiological importance of the purinergic component of sympathetic vasoconstrictor nerves is difficult to establish in vivo. Species differences and differences between vascular beds within a given species complicate the situation, making generalizations difficult to apply. There may be a greater purinergic component in small as opposed to large vessels.9,10
Neuropeptide Y (NPY) is a 36 amino acid peptide which, in addition to its presence in sympathetic vasoconstrictor nerves, is also found in areas of the brain involved in cardiovascular control and may play an important role in modulating the sympathetic nervous control of blood pressure.11 NPY is preferentially released by high-frequency stimulation of perivascular nerves and has both direct vasoconstrictor effects (mediated via smooth muscle Y1 receptors) and potentiates the action of noradrenaline and ATP. These vasoconstrictor actions of NPY are most evident in vessels with wide neuromuscular junctions (i.e. larger vessels).
The concept of cotransmission in sympathetic vasoconstrictor nerves and other vasomotor nerves (see Table 2.1) provides the potential for greater complexity and fine tuning of control via multiple interactions between neurotransmitters. Actions at the presynaptic nerve membrane should also be mentioned here, as neuromodulatory actions occur often via receptors which differ from those on the postsynaptic membrane.12 The complexity of this system is illustrated in Table 2.2, which lists some of the receptors found on the presynaptic membrane of sympathetic vasoconstrictor nerves.
Table 2.2 Presynaptic receptors which modulate sympathetic vasoconstrictor nerves
| RECEPTOR | ENDOGENOUS LIGAND | NEUROMODULATORY EFFECT |
|---|---|---|
| α2-Adrenoceptors | Noradrenaline, adrenaline | Inhibits transmitter release |
| P1-Purinoceptors | Adenosine (formed from ATP) | Inhibits transmitter release |
| Y2-Receptors | NPY | Inhibits transmitter release (small vessels particularly) |
| DA2-receptors | Dopamine | Inhibits transmitter release |
| β2-Adrenoceptors | Adrenaline (circulating hormone) | Facilitates transmitter release |
| AT1-receptors | Angiotensin II (circulating hormone) | Facilitates transmitter release |
| 5-HT3-receptors | 5-HT released from platelets or from perivascular nerves | Facilitates transmitter release |
Drugs which influence vasomotor nerve function
Most drugs have been developed to target the sympathetic vasoconstrictor nerves, as the therapeutic aim has been to reduce vascular resistance and increase venous capacitance, either in the management of hypertension or congestive heart failure. Antagonists with selectivity for α1-adrenoceptors have proved to be the class which produce the least side effects. Nonselective α-adrenoceptor antagonists were less effective and caused more reflex tachycardia because blockade of presynaptic α2-adrenoceptors increased the release of noradrenaline at all sympathetic postganglionic nerve synapses. Alpha1-adrenoceptor antagonists are effective at reducing vasomotor tone initially. Indeed, a first-dose severe hypotensive effect can be a problem. With repeated dosing, however, the effect is reduced. This may well be due to physiological tolerance, via activation of other endogenous vasoconstrictor systems, which might include increased importance of the cotransmitters (ATP and NPY) utilized by these nerves.13 Drugs which inhibit the effects of ATP and NPY at their respective vascular receptors are under development but are not yet sufficiently well understood or developed for clinical use to be contemplated.14–16
Nervous control of cardiac function
By contrast, the distribution of cardiac sympathetic nerves is much more widespread, allowing this system to influence not only heart rate but also the inotropic state of cardiac muscle. Resting cardiac sympathetic tone in equine hearts is thought to be extremely low. The original concept that noradrenaline acts exclusively on β1-adrenoceptors to mediate the effects of sympathetic stimulation, namely increased heart rate and force of contraction, is too simplistic. Although β1-adrenoceptors predominate in all species including the horse,17 β2-adrenoceptors are found in the myocardium, particularly in atria and nodal tissue in man, rabbit and cat18 and horse,19 and probably explains why drugs with mixed β1– and β2-adrenoceptor agonist actions (e.g. isoprenaline) increase heart rate more effectively than those selective for β1-adrenoceptors (e.g. dobutamine). In some species, there is good evidence that with chronic high levels of stimulation, the proportion of β2-adrenoceptors in relation to β1-adrenoceptors increases, while the overall number of β-adrenoceptors and their efficiency of coupling to cyclic AMP production decreases.20 This phenomenon is less clear in the horse, and in 10 horses with congestive heart failure the density of β-adrenoceptors was unchanged and the density of β2-adrenoceptors was increased only in five horses.17 Beta-adrenoceptors have been implicated in cardiac remodelling leading to left ventricular enlargement and the β-adrenoceptor antagonists have been shown to reverse these effects in both pressure and volume loading.21–25 As such, this class of agent may have potential therapeutic value in the horse with cardiac hypertrophy. In addition, α1-adrenoceptors can be demonstrated in cardiac muscle. Stimulation of these receptors is thought to contribute, in a small way, to the positive inotropic effect of sympathetic nerve stimulation (with no effect on heart rate), but this contribution may increase in importance in disease states, when downregulation of the β1-adrenoceptors occurs, and may also increase the tendency for dysrhythmias to develop in hearts driven by high sympathetic tone.18 Alpha1-adrenoceptors have also been implicated in the development of myocardial fibrosis during cardiac remodelling and offer potential targets for therapeutic intervention23 although there is no clinical evidence to support their use in this way in any species.
HORMONAL CONTROL OF THE CARDIOVASCULAR SYSTEM
The key hormones currently thought to be involved in regulation of the cardiovascular system are summarized in Table 2.3. It is not possible to review these systems in any detail here, and areas with potential therapeutic importance will be focused on.
Table 2.3 Hormones of key importance in the control of the cardiovascular system
The renin–angiotensin–aldosterone system
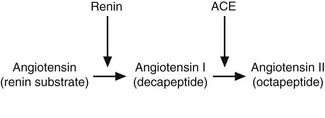
The kidney responds to reduced perfusion, reduced solute filtration and increased sympathetic nerve stimulation by producing an enzyme, renin, which is secreted by modified smooth muscle cells of the afferent arterioles in the juxtaglomerular apparatus.26 Renin then acts on its circulating substrate to produce angiotensin I, which is converted to the active hormone angiotensin II by the enzyme angiotensin-converting enzyme (ACE) present on endothelial cells, particularly in the lung (Fig. 2.1).
This is the conventional view of the way angiotensin II is produced, where it is thought to function in an endocrine fashion as a defence against hypovolaemia, hypotension and circulatory shock. There is good evidence for the local presence of renin (or renin-like enzyme activity), angiotensinogen and ACE, leading to local production of angiotensin II, in a number of tissues, including the myocardium, kidney and brain.27 In the myocardium, upregulation of the system is thought to occur in response to increased ventricular wall stress28 and may be important in the development of cardiac enlargement and fibrosis.29–33 The effects of angiotensin II are summarized in Table 2.3. Aldosterone is synthesized in response to angiotensin II and acts via mineralocorticoid receptors to preserve sodium by inducing reabsorption from the distal nephron. This results in an increase in circulating volume, and an increase in preload in the compensated diseased heart. However, volume overload ultimately leads to volume-induced hypertrophy, myocardial remodelling and fibrosis34 and therefore inhibition of the actions of aldosterone, either by blocking production or its receptors, could reduce progression of cardiac disease.
Aldosterone has been proposed as a biomarker of severity of cardiac failure since its concentrations correlate with severity of disease in human patients,35 although not in dogs with mitral valve regurgitation.36,37 In the horse, increased serum concentrations of aldosterone have been reported in association with valvular regurgitation of the mitral, aortic and tricuspid valve.38 Although this study failed to differentiate the underlying disease it did show higher aldosterone concentrations in animals with significant left ventricular enlargement. Some horses with both atrial and ventricular enlargement had aldosterone concentrations similar to normal horses and in our laboratory horses with aortic valve regurgitation had no increase in serum activities of plasma renin, concentrations of angiotensin II or aldosterone (unpublished observations). It is not clear where these differences in study results arise although this may suggest that in certain forms of cardiac disease in the horse activation of the renin–angiotensin system is not important. Indeed, horses with compensated aortic valve regurgitation have increased systolic pressure due to increased end-diastolic volume (preload), such that there is no stimulation of renin release from the kidneys since systolic pressures are normal.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


