Chapter 19 Heart failure
INTRODUCTION
Heart failure is the pathophysiological state in which an abnormality of cardiac function is responsible for failure of the heart to fill with or eject blood sufficient to meet the metabolic requirements of the tissues1,2 (see Chapter 5). Failure of the myocardium is usually present, however the initiating pathology may be elsewhere within the heart, for example, involving the endocardium, pericardium or great vessels (see Chapter 4). Historically, medical and veterinary students were taught to consider heart failure as having two forms: forward and backward failure. These theories focused on cardiovascular haemodynamics and postulated that clinical signs of heart failure could be attributed to either failure of the cardiac pump (forward failure) or damming up of blood behind one or both ventricles (backward failure).2 These concepts are now outmoded and it is clear that heart failure is a complex process involving not only structural abnormalities and haemodynamic mechanisms but also neuroendocrine, biochemical and genetic pathways through which either an increased haemodynamic burden or a reduction in myocardial oxygen delivery leads to abnormal myocardiac structure and function.1,2 Although not yet fully investigated the autonomic nervous system is activated in horses with heart failure3 and there is also some evidence supporting activation of the renin–angiotensin activating system, and plasma aldosterone concentrations rise as the severity of valvular disease increases.4 Initially, these neuroendocrine adaptations have beneficial effects in maintaining cardiac output, but ultimately they create deleterious effects on the heart, and also on the vasculature and organs such as the kidney1,2,5 (see Chapters 2 and 4).
CLINICAL SIGNS OF HEART FAILURE
The clinical signs of heart failure can be attributed to a combination of reduced cardiac output and increased ventricular filling pressures. Increased ventricular filling pressures can lead to signs of congestion affecting one or both sides of the heart. These processes occur in combination to varying degrees in most of the cardiac conditions that lead to heart failure, and provided that oversimplification is avoided, it can be helpful for the clinician to differentiate the signs of reduced cardiac output from congestion of each side of the circulation as this may lead to the formation of the most accurate list of differential diagnoses. The term congestive heart failure (CHF) should be reserved for those cases showing left, right or biventricular venous congestion and it is important to recognize that the term heart failure not only encompasses CHF but also includes cases in which the ventricle fails and cardiac output drops without development of congestion. The clinical consequences of reduced cardiac output include tachycardia, weight loss, weakness, exercise intolerance, pale mucous membranes, weak arterial pulses, ataxia and syncope. Reduced renal output is common and increases in serum creatinine concentration arise through both prerenal and direct renal mechanisms.6 ( PC, VMD)
PC, VMD)
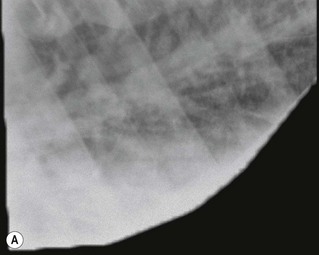
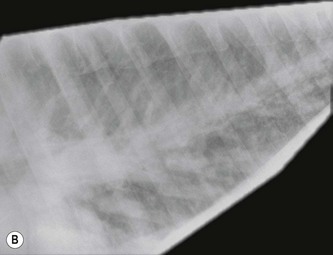
Increased ventricular filling pressure leads to congestion of the systemic, the pulmonic or both sides of the circulation and the clinical signs of CHF. The exact range of signs will depend on the specific causative lesion and its location. Lesions that cause the left heart to fail, such as severe mitral insufficiency (MI) and large ventricular septal defects, lead to congestion of the pulmonary circulation.6,7 The distribution of fluid between the interstitium and the plasma is dependent on the balance between oncotic pressure and hydrostatic forces. The hydrostatic forces rise in response to increased ventricular filling pressure and pulmonary venous engorgement in heart failure, forcing fluid from the pulmonary capillaries into the pulmonary interstitium faster than the lymphatics can remove it, to create pulmonary oedema. Oedema fluid is relatively low in protein and as it moves from the pulmonary capillaries into the interstitium, the protein is returned to the blood, raising the plasma oncotic pressure and lowering the interstitial oncotic pressure and thereby counteracting the tendency for fluid to leave the circulation under hydrostatic forces. Therefore, pulmonary oedema can be self-limiting for a considerable period of time.8 In slowly progressive lesions, early or mild heart failure, raised respiratory rates, particularly after exercise, and crackles and moist bronchovesicular sounds may be the only clinical signs of pulmonary oedema.8 Thoracic radiographs demonstrate an interstitial pattern and pulmonary venous congestion. If pulmonary capillary pressure rises acutely, the oncotic pressure within the interstitium rises rapidly to produce alveolar flooding.8 With a more acute onset or in advanced heart failure, there may be dyspnoea, coughing and profuse nasal discharge which is typically white or pink-tinged and frothy (Fig. 19.1) and thoracic radiographs demonstrate a fluffy alveolar pattern (Fig. 19.2). Pulmonary hypertension can also lead to dilatation and eventually rupture of the pulmonary artery6,9,10 (see Fig. 18.4). Rupture of the pulmonary artery is not necessarily immediately fatal and there may be a history of episodes of syncope and distress on several occasions before the horse dies (see Chapter 18). ( AF, IE)
AF, IE)

Right ventricular volume overload is less common as an isolated state. However, clinical signs of left heart failure may frequently go unnoticed, with slowly progressive disease until the right heart fails in response to pulmonary hypertension.6 Clinical signs of right-sided CHF include jugular distension, pulsations of the jugular veins extending beyond the normal distal one-third of the neck, distension of other peripheral veins such as the lateral thoracic veins, and ventral, muzzle, preputial and limb oedema. Ascites is difficult to appreciate on physical examination in horses, but ultrasonography may reveal increased volumes of fluid in the abdominal cavity and congestion of the hepatic vessels. Horses also frequently develop pleural effusions with heart failure which can be demonstrated by absence of respiratory sounds over the ventral lung field and percussion of ventral thoracic dullness, and which can be visualized radiographically or ultrasonographically. ( ACF, PC, VMD)
ACF, PC, VMD)
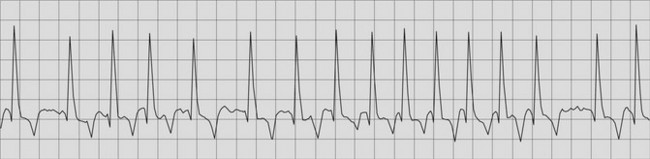
Many of the lesions associated with heart failure lead to enlargement of either the left or right atrium. This predisposes the horse to the development of atrial fibrillation, an extremely common finding in CHF11 (Fig. 19.3). It is important to differentiate horses in which atrial fibrillation is a consequence of heart failure rather than a primary condition, because as discussed in Chapter 13, quinidine sulphate is indicated in lone (uncomplicated) atrial fibrillation and contraindicated in CHF.11 A heart rate of greater than 60 bpm, loud cardiac murmurs and echocardiographic evidence of valvular or other cardiac lesions suggest CHF is present in horses with atrial fibrillation (see Fig. 19.3). ( AF)
AF)
DIFFERENTIAL DIAGNOSIS OF HEART FAILURE
The prevalence of CHF in horses is unknown and is likely to vary considerably between different horse populations. In one study examining causes of death among a group of 1153 horses the majority of which were middle-aged or older, there were no cardiac-related deaths in horses less than 17 years of age, and 5% of deaths in the 15–23 years age group and 8.5% of deaths in the >24 years age group were considered to relate to cardiac disease.12 In the author’s cardiological practice serving a wide range of racing, sports, pleasure and breeding horses of all ages, approximately 2% of cardiac admissions have CHF.
Disorders affecting a variety of body systems can present in a similar manner to CHF. In addition to CHF, the main differential diagnoses for horses presenting with peripheral oedema can be broadly classified: as (1) conditions associated with protein loss, such as protein-losing enteropathy; (2) conditions associated with obstruction to lymphatic drainage, such as lymphoma or lymphadenopathy; and (3) peripheral vasculitis such as purpura haemorrhagica. In horses presenting with mild to moderate respiratory disease, there are numerous possible conditions that must be considered while with acute severe pulmonary disease, in addition to left-sided heart failure, conditions such as interstitial pneumonia and acute lung injury must be included in the list of differential diagnoses. The clinical signs associated with low cardiac output are nonspecific and myocardial failure must be differentiated from other conditions associated with circulatory failure (i.e. shock) such as abdominal catastrophes and haemorrhage (see Chapter 21).
Causes of heart failure in the horse are listed in Table 19.1. The majority of these diseases can also present at less advanced stages and are described in more general terms elsewhere in this book. The reader is advised to consult specific chapters for additional details (see Chapters 13–18). The aim of this section is to assist the clinician in devising an appropriate diagnostic and therapeutic plan when presented with a horse showing signs of heart failure.
Table 19.1 Conditions that commonly cause heart failure in the horse
Predominantly left-sided or biventricular failure Predominantly right-sided failure |
Biventricular CHF is most commonly seen in association with severe, acquired disease of the aortic and/or mitral valves.6,13–18 However, occasionally horses with large ventricular septal defects and other congenital cardiac defects can present with left-sided or bilateral CHF.7,19 Primary right-sided CHF is less common but occasionally occurs with severe tricuspid (TI) or pulmonic insufficiency, congenital disease and cor pulmonale.20,21 Pericarditis causes heart failure because cardiac tamponade due to effusion within the pericardial sac or loss of elasticity in restrictive pericarditis increases the intrapericardial pressure and prevents diastolic filling. This process affects both sides of the heart, but typically signs associated with the systemic circulation predominate because the lower pressures within the right side of the circulation can more readily be overwhelmed.22–25 Rapid tachycardia can also impede diastolic function sufficiently to induce signs of heart failure by decreasing the time available for the heart to fill in diastole.1 In this situation, signs of left-sided congestion predominate.
DIAGNOSTIC APPROACH IN HEART FAILURE
Echocardiography is an extremely valuable diagnostic aid when cardiac failure is suspected as both cardiac structure and function can be assessed with this imaging modality.26–29 Echocardiography can be useful, in particular in identifying chamber dilation and compromised myocardial function. In compensated left ventricular volume overload, the fractional shortening and ejection fraction should be increased and where normal or decreased fractional shortening and ejection fraction are detected in the presence of volume overload, ventricular remodelling and loss of myocardial function should be suspected (see Chapter 9). Regardless of cause, typical echocardiographic features of generalized myocardial dysfunction include ventricular dilatation, hypomotility, increased septal–mitral E-point separation, reduced fractional shortening and reduction in the movement of the aortic root on M-mode echocardiography. ( AF, PC, VMD) There is often mitral insufficiency secondary to valve annulus dilatation. Where the myocardial dysfunction has arisen as a consequence of valvular insufficiency and volume overload, the left ventricular free wall is often more obviously affected than interventricular septum. (
AF, PC, VMD) There is often mitral insufficiency secondary to valve annulus dilatation. Where the myocardial dysfunction has arisen as a consequence of valvular insufficiency and volume overload, the left ventricular free wall is often more obviously affected than interventricular septum. ( VMD)
VMD)
Direct measurement of pulmonary artery and capillary wedge pressure is the most reliable method for documenting pulmonary hypertension. ( PH) Radiology can be limited in its ability to demonstrate milder forms of oedema and clinical signs do not necessarily relate closely to the degree of oedema visible radiographically. Nevertheless, radiography is frequently the most practical means of identifying pulmonary oedema and evaluating the response to therapy (see Fig. 19.2). Severe pulmonary oedema can be visualized with diagnostic ultrasonography as multifocal areas of hypoechoic, nonaerated lung at the periphery (Fig. 19.4). However, pulmonary oedema is most severe at the hilar area and often does not always extend to the periphery of the lung where it can be visualized with ultrasonography. Diagnostic ultrasonography is more useful in documenting the presence of pleural effusion, hepatic congestion and ascites. (
PH) Radiology can be limited in its ability to demonstrate milder forms of oedema and clinical signs do not necessarily relate closely to the degree of oedema visible radiographically. Nevertheless, radiography is frequently the most practical means of identifying pulmonary oedema and evaluating the response to therapy (see Fig. 19.2). Severe pulmonary oedema can be visualized with diagnostic ultrasonography as multifocal areas of hypoechoic, nonaerated lung at the periphery (Fig. 19.4). However, pulmonary oedema is most severe at the hilar area and often does not always extend to the periphery of the lung where it can be visualized with ultrasonography. Diagnostic ultrasonography is more useful in documenting the presence of pleural effusion, hepatic congestion and ascites. ( PC)
PC)
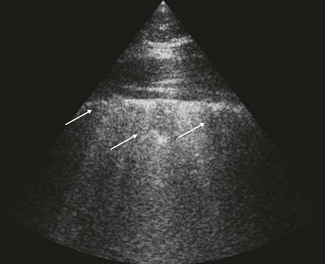
Figure 19.4 A thoracic ultrasonogram in an 11-year-old Arabian mare with acute myocarditis and pulmonary oedema (see Fig 19.1). The periphery of the lung is irregular with hypoechoic nonaerated areas representing oedema (arrows) and the lung has lost the horizontal reverberation pattern that is seen in normal aerated lung.
The identification of myocardial disease and dysfunction is an important component of assessment of horses with heart failure. Electrocardiography is required to demonstrate arrhythmias that may accompany heart failure, such as atrial tachycardia, atrial fibrillation, supraventricular or ventricular premature depolarizations, ventricular tachycardia or pre-excitation syndrome. Ambulatory ECG techniques are particularly useful in demonstrating paroxysmal arrhythmias and documenting their frequency over prolonged periods (see Chapters 6 and 10). Novel biomarkers are currently being assessed in horses and these are discussed in more detail in Chapter 12. Cardiac troponin may be increased in some cases6,30 but it is important to recognize that a normal cardiac troponin concentration does not rule out the presence of myocardial pathology, particularly myocardial fibrosis.
SPECIFIC FORMS OF HEART FAILURE
Left-sided valvular insufficiency( AF, VMD)
AF, VMD)
Degenerative valvular disease of the aortic valve is extremely common in horses12,31,32 (see Chapter 16). In the majority of horses with aortic insufficiency (AI), the lesion is well tolerated and the proportion of affected individuals that progress to develop CHF is unknown: in one study, 86% of horses with AI showed no signs of heart disease at rest while 13.2% had signs of CHF at rest, however, this population was drawn from an elderly population and horses that develop CHF with AI are typically in their late teens or older. Clinical signs of hyperkinetic pulses and a widely radiating diastolic murmur are consistent with severe AI.13 With echocardiography, nodular lesions of the aortic valve and large regurgitant jets are visualized.13,14,28,33 The onset of myocardial failure often appears to herald the onset of clinical signs of CHF; in horses with well-compensated but severe AI, the left ventricle is usually dilated but hyperkinetic13,14 (Fig. 19.5). Left ventricular volume overload increases end-diastolic left ventricular diameter and causes the ventricle to adopt a globoid shape with a rounded apex.13 In the hyperkinetic ventricle, septal and left ventricular free-wall movement is exaggerated and the fractional shortening is increased.13 As the ventricle begins to fail, myocardial contractility is often depressed. Fractional shortening, which is influenced by a variety of factors including myocardial contractility, will decrease and subjectively the ventricle may appear to be hypomotile (see Fig. 19.5). Most horses that develop overt CHF with degenerative AI have ventricular dilatation to a degree that can disrupt the mitral valve annulus and cause MI (Fig. 19.6). In fact, it is the onset of MI that may be the event that has ultimately caused the cardiac decompensation and horses with multiple murmurs are more likely to go into CHF than those with AI alone.3 Therefore, the development of a murmur of MI in a horse with previously well-compensated AI should alert the clinician to the possibility of progression of the disease. These horses typically have end-diastolic left ventricular diameters in excess of 16 cm (see Fig. 19.5).
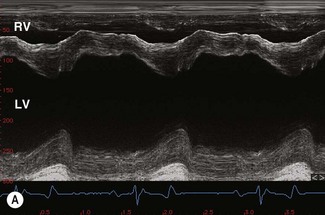
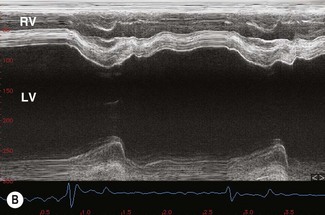
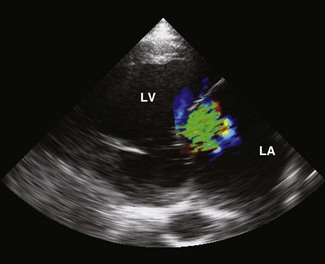
Figure 19.6 A left parasternal long-axis echocardiogram of the left atrium (LA) and ventricle (LV) from the horse illustrated in Fig. 19.5B. The mitral valve annulus is dilated and there is a large jet of mitral regurgitation (green). The LA has an increased diameter of 18.9 cm (normal <13.5 cm).
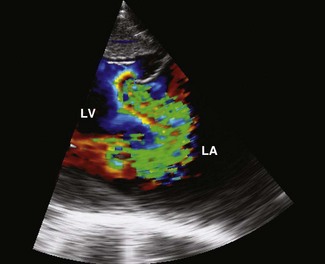
Severe MI can be caused by valvular degeneration or inflammation6,15,31 and may also arise secondary due to ventricular dilation as a result of myocardial, congenital or aortic disease. In one study of horses with severe MI, almost 50% of affected horses had evidence of concurrent myocardial disease.6 Severe MI is seen in a wider range of age groups than AI. Auscultation reveals a loud pan- or holosystolic murmur radiating over a wide area with severe MI and the third heart sound is often loud because there is increased ventricular filling in diastole.6 In chronic degenerative cases, nodular thickening of the valve may be visible echocardiographically and subsequently on post-mortem examination. Severe mitral regurgitation occupies the majority of the left atrium, has a wide base, may originate from more than one source at the valve leaflets and may demonstrate proximal flow convergence29 (Fig. 19.7). Severe MI is poorly tolerated and consequences include atrial enlargement (see Fig. 19.6), atrial fibrillation (see Fig. 19.5), ventricular tachyarrhythmias, pulmonary oedema (see Fig. 19.4) and, occasionally, pulmonary artery dilatation and rupture6,15–18 (see Fig. 18.4). Dilatation of the left atrium can be appreciated most readily from two-chamber left parasternal images (see Fig. 19.6), and in mature horses with severe MI, left atrial diameters of 16.2–23 cm have been reported, normal being less than 13.5 cm.6,29 The normal ratio of the pulmonary artery measured in long-axis to the aortic root diameter measured in long-axis is around 0.8. Dilatation of the pulmonary artery is diagnosed if its diameter is greater than that of the aorta (Fig. 19.8).6,9 In many horses with pulmonary artery dilatation, pulmonic valvular insufficiency is present (see Fig. 19.8), again probably the result of pulmonary hypertension.6 Horses with severe MI also often have reduction in the aortic root diameter due to reduced cardiac output (see Fig. 19.8). In one report describing horses with severe mitral valve disease, 64% of horses had pulmonary artery diameters which exceeded that of the aorta by greater than 1 cm.6
< div class='tao-gold-member'>
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


