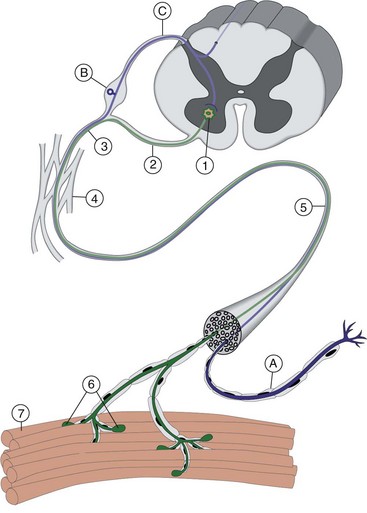
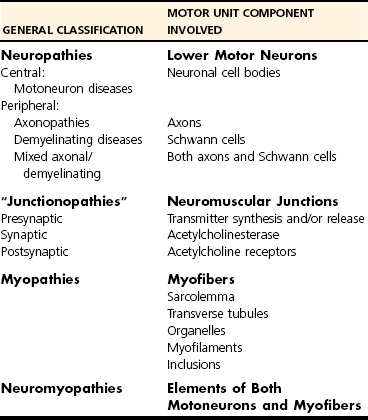
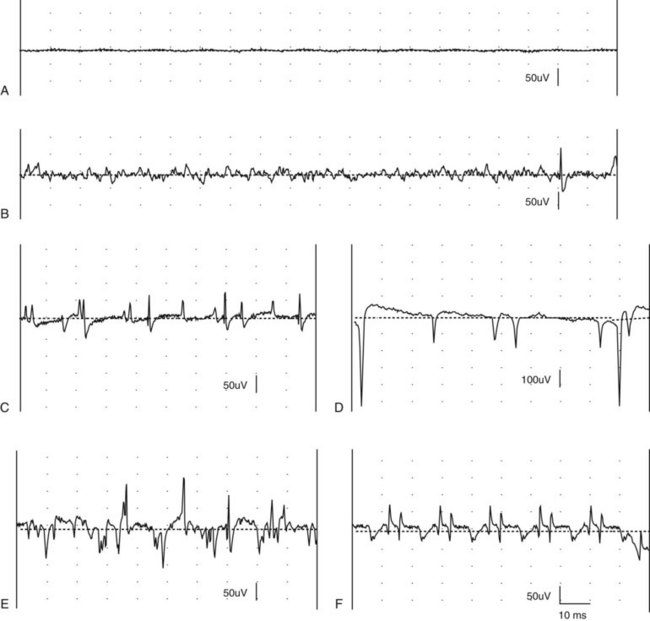
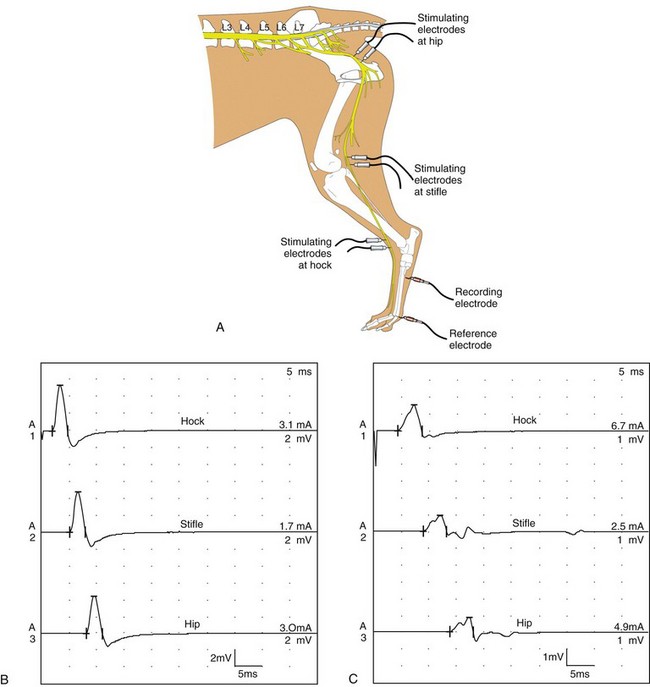
Chapter 27 Neuromuscular diseases are directed against, and are limited to, one or more subdivisions of the motor unit (i.e., the basic functional and anatomic organization of neurons and muscle fibers). These diseases include neuropathies (disorders of the neuron, including the cell body, axon, Schwann cell, and myelin), “junctionopathies” (disorders of neuromuscular transmission), myopathies (disorders of muscle fibers), and neuromyopathies (disorders of both neurons and muscle fibers). Diagnosis of neuromuscular disease is dependent on thorough physical and neurologic examinations, specific serologic testing, electrophysiologic evaluation, and examination of optimally processed muscle and peripheral nerve biopsies.19,27,53 1. To determine whether the animal’s presenting complaint and physical findings are the result of a neuromuscular disorder, or are due to some other cause (e.g., orthopedic disease). 2. To formulate a complete list of differential diagnoses that will lead to proper identification of the location and cause of the neuromuscular disorder. 3. To institute appropriate therapy (pharmacologic if available) and supportive care to modify the basic disease process and improve the animal’s quality of life. Management of an animal suspected to have a neuromuscular disease may be extremely frustrating, as there is a narrow range of presenting clinical signs for the numerous disorders affecting the neuromuscular system. These clinical signs frequently are common to many different disorders, regardless of the cause.18 Each motor unit is composed of a single motoneuron and a variable number of skeletal myofibers innervated by that motoneuron (Figure 27-1).23 Essential components of each motor unit include the following: 1. A motoneuron consisting of its cell body (located within the central nervous system, either in the cranial nerve nuclei of the brainstem or in the ventral horns of gray matter in the spinal cord) and its peripheral axon, supported by Schwann cells. All motor units are not alike and may vary with regard to the following: 1. Size (the number of myofibers innervated by a single motoneuron). 2. Histochemical properties of the myofibers. 3. Functional properties related to the speed of contraction and resistance to fatigue. At least three and possibly five basic types of motor units have been identified, based on their contractile and histochemical properties.23 The myofiber-type composition of motor units is homogeneous. Each motor unit is composed of a single histochemical myofiber type, and not a mixture of myofiber types. Individual muscles usually are composed of a mixture of motor unit (myofiber) types, and the relative proportions of each may vary considerably between muscles. Individual myofibers of each motor unit are uniformly distributed throughout a relatively large area in a muscle, and myofibers of the same motor unit rarely are contiguous. This scattered distribution results in a mosaic pattern of myofiber types within transversely sectioned and stained muscles. For a given muscle within the same species, the myofiber-type composition and the mosaic distribution pattern appear to be reasonably constant and characteristic for that muscle. A useful classification scheme for neuromuscular diseases is based on the anatomic motor unit components that are primarily involved in the pathogenesis of the muscle weakness (Table 27-1). Using this classification, neuromuscular diseases are broadly subdivided as follows: Dysfunction of the motor unit results in lower motor neuron signs, seen clinically as muscle weakness (Box 27-1). Expression of this weakness may vary considerably and may include lameness, paresis/paralysis, gait abnormalities, exercise-related weakness, dysphagia, regurgitation, dyspnea, and dysphonia. The distribution of involvement may be local, regional, or generalized. In addition, there may be gross deformities of muscle mass (i.e., atrophy, hypertrophy, and skeletal deformities). Any patient presenting with some form of clinical weakness should be viewed as potentially having a motor unit disorder. The conclusion that the patient is “merely weak because it is sick” should not be readily assumed without meticulous evaluation of the motor unit. Braund has described a myopathic syndrome and a neuropathic syndrome to aid in the recognition of neuromuscular disorders.11 The principal signs of myopathic syndrome are generalized weakness, exercise intolerance, stiff and stilted gait, body/limb tremors, localized or generalized muscle atrophy, localized or generalized muscle hypertrophy, “percussion dimple” contracture, apparent muscle pain on palpation, limited joint movement (e.g., contracture), regurgitation or altered esophageal motility (megaesophagus), ventroflexion of the head and neck, and trismus.11 The principal signs of motor neuropathic syndrome are flaccid paresis/paralysis of innervated structures (e.g., limb/facial muscles, esophagus, larynx, anal sphincter),31 neurogenic muscle atrophy, reduced/absent reflexes and muscle tone, and muscle fasciculations; the principal signs of sensory neuropathic syndrome are decreased pain response (hypalgesia) or sensation (hypesthesia), proprioceptive deficits, abnormal sensation/sensitivity (paresthesia) of the face, trunk, or limbs, self-mutilation, and reduced/absent reflexes in the absence of muscle atrophy.11 Signs of autonomic dysfunction may be seen alone or in combination with sensorimotor neuropathy. The principal signs of autonomic neuropathy are anisocoria or dilated pupils, decreased tear secretion, decreased salivation, and bradycardia.11,40 Establishing a diagnosis requires an informed and coordinated approach to defining a problem list through associations and direct observations (i.e., a diagnostic plan).27 Essentials of the diagnostic plan are summarized in Box 27-2. Box • 27-2 Diagnostic Plan for Neuromuscular Diseases 3. Physical examination (including orthopedic examination) 5. Minimum database—not specific for neuromuscular disease 6. Minimum database—specific for neuromuscular disease 7. Specific diagnostic procedures Specific laboratory testing may be required for the diagnosis of neuromuscular disease.50 In addition to a minimum database that is not specific for neuromuscular disease, a minimum database specific for neuromuscular disease may include the measurement of serum creatine kinase activity, serum electrolyte determination, blood or plasma lactate concentration, urine testing for myoglobinuria, thyroid screening, or serum assay for acetylcholine receptor antibody.50 Elevated creatine kinase is a sensitive indicator of skeletal muscle damage, although creatine kinase activity within the normal range does not rule out inflammatory muscle disease.3 Large elevations in creatine kinase (>10,000 IU/L) may be seen in association with muscular dystrophies, necrotizing myopathies, and inflammatory myopathies. Moderate to marked creatine kinase elevations may be seen in anorectic cats.24 Mild to moderate creatine kinase elevations (<10,000 IU/L) may be seen in inflammatory myopathies and in some degenerative myopathies. Electrolyte alterations can result in muscle weakness and may be found in endocrine and metabolic diseases and in inherited genetic diseases, including hyperkalemic and hypokalemic periodic paralysis.42 Lactic acidosis and exercise intolerance syndromes are known to be associated with inborn metabolic errors. Lactate concentration should be measured at rest and following 10 minutes of exercise (intensity determined by clinical status of the patient).55 Lactate concentrations also should be evaluated in an age- and size-matched control dog exercised at a similar intensity. In addition to metabolic myopathies, lactic acidosis may occur with excitement during venipuncture or with abnormalities of ventilation and perfusion. In addition to plasma lactate determinations, evaluation of resting and post-exercise plasma pyruvate concentrations may be performed in animals suspected to have a mitochondrial myopathy.42 Following significant muscle injury (i.e., rhabdomyolysis), myoglobinuria may be observed as “coca-cola”–colored urine.54 Once hematuria has been excluded by examination of the urine sediment, pigmenturia due to myoglobin may be distinguished from that due to hemoglobin by an ammonium sulfate precipitation test that is available through most commercial laboratories. Hypothyroidism may be associated with muscle weakness in dogs. Of clinical significance, marked myopathic features of hypothyroidism can be found in muscle biopsies of hypothyroid dogs without clinical signs of weakness, suggesting that hypothyroid myopathy may be an underrecognized complication.45 Such dogs may escape clinical detection if not subjected to vigorous exercise regimens, as myopathy may exist in the absence of weakness or abnormal biochemical markers of muscle damage commonly used in the clinical setting. Weakness may be a clinical feature of hyperthyroidism in cats and can be diagnosed easily by appropriate clinical signs and evaluation of thyroid function (i.e., total thyroxine [T4]). Autoimmune (acquired) myasthenia gravis may occur in many clinical forms.49,51 Because the spectrum of clinical presentations is broad and variable, autoimmune myasthenia gravis should be high on the list of differential diagnoses for any dog or cat presenting with focal or generalized neuromuscular weakness, megaesophagus, or dysphagia. The acetylcholine receptor antibody test remains the “gold standard” for the diagnosis of acquired myasthenia gravis. Additional tests may be indicated, based on results of the neuromuscular minimum database or after pathologic changes in muscle biopsies have been determined. For example, in addition to clinical signs, definitive diagnosis of masticatory myopathy can be made with a positive 2M-antibody titer. Further laboratory diagnostics may include testing for infectious agents such as Toxoplasma gondii, Neospora caninum, or tick-related diseases; evaluation of a metabolic panel, including quantitative plasma amino acids and urinary organic acids; blood gas and anion-gap analysis; and quantification of muscle, plasma and urine carnitine concentrations.50 Prevention of inherited disease in companion animals is largely dependent on prebreeding identification of carriers of autosomal recessive disease traits. Molecular diagnosis is emerging as a convenient and reliable method of carrier detection, but few molecular diagnostic tests of inherited neuromuscular disease are readily available. New test development depends on investigations to identify disease genes and disease-causing mutations.26 Two groups of diagnostic tests are specific for neuromuscular diseases: (1) electrodiagnostic testing,16,21,32 and (2) muscle/nerve biopsy.19 Electromyography: Electromyography is the detection and characterization of electrical activity (potentials) recorded from a patient’s muscles.32 A systematic study of individual muscles permits accurate determination of the distribution of muscles affected by neuromuscular disorders. Electromyography is routinely performed as part of a complete neuromuscular workup that also may include nerve conduction velocity determinations (both motor and sensory), repetitive nerve stimulation, and late wave testing (F-waves, H-reflexes), along with muscle and nerve biopsy collection.16 In addition, electromyography may be utilized in conjunction with imaging techniques (e.g., computed tomography [CT], magnetic resonance imaging [MRI]) when nerve root or central nervous system involvement is suspected. In small animals, electromyography is typically conducted with the muscles at rest (i.e., not contracting) and usually are done with the patient under general anesthesia. Under these conditions, resting potentials across muscle fiber membranes are maintained, and hence, normal muscles at rest are “electrically silent” (Figure 27-2, A). However, with depolarization of muscle fiber membranes, potentials are generated that have a characteristic waveform, usually consisting of negative and positive phases. The potentials generated are evaluated for amplitude, duration, number of phases, polarity, frequency, and repetition. “Normal” Spontaneous Activity: Four types of spontaneous electrical activity may be detected in normal muscle: insertional activity, miniature end-plate potentials, end-plate spikes, and motor unit action potentials. Insertional activity: Brief bursts of electrical activity (potential changes) are induced by insertion of the needle electrode into the muscle. This activity results from irritation (mechanical disruption or stimulation) of single muscle fibers as the needle electrode is advanced. Normal muscles become “electrically silent” immediately upon cessation of needle electrode movement. Increased or prolonged insertional activity (whereby insertional activity continues for a short time after the needle electrode has stopped moving) may be observed in neuromuscular diseases. Affected muscle fibers are hyperexcitable, having a lowered threshold owing to diminished membrane potentials. Prolonged insertional activity may be the only abnormal finding early in the course of a neuromuscular disease. Its opposite, decreased insertional activity, is an indication that significant (or complete) loss of myofibers has occurred and may be the only electromyographic finding in muscles with end-stage disease (although it is often subtle and easily missed). Miniature end-plate potentials: Miniature end-plate potentials may be seen when the needle electrode is positioned in proximity to a neuromuscular junction. This activity is also referred to as end-plate noise. These potentials result from localized muscle membrane depolarization that is induced by the normal ongoing release of individual quanta of acetylcholine from the nerve ending (see Figure 27-2, B). End-plate spikes: Occasionally, a single normal myofiber may depolarize completely, resulting in an action potential (or “end-plate spike”). It is important to distinguish end-plate spikes from abnormal potentials (such as fibrillation potentials) (see Figure 27-2, B). Motor unit action potentials: Motor unit action potentials may be recorded from normal muscle that is not completely at rest (e.g., during a voluntary muscle contraction). Motor unit action potentials represent a compound action potential of myofibers within the recording range of the needle electrode. They may increase in amplitude and number with increased activation of the same or new motor units (“recruitment”), respectively. Abnormal Spontaneous Activity: Abnormal spontaneous activity occurs when a stationary needle electrode is positioned in proximity to an abnormal myofiber or group of myofibers. Stimulation is not applied to the muscle, other than insertion of the electrode. Three patterns of abnormal spontaneous activity may be seen on electromyography: fibrillation potentials (or “fibs”) and positive sharp waves (or “sharps”), complex repetitive discharges, and myotonic potentials (see Figure 27-2, C through F). A standardized rating system is used to grade spontaneous activity in each muscle tested. Muscle activity is graded from normal (0) to severe (4+). “Fibs” and “sharps”: These abnormal potentials represent the same underlying pathologic changes and differ in morphologic findings only in relation to their orientation to the recording electrode at the time of discharge. Both potentials arise from spontaneously firing, hypersensitive, single myofibers as a result of destabilization of the sarcolemmal membrane. Fibs and sharps may occur secondary to denervating diseases (neuropathies), or as a consequence of a primary myopathic change (myopathies) (see Figure 27-2, C through E). Complex repetitive discharges: Complex repetitive discharges are polyphasic repetitive waveforms with a uniform frequency, configuration, and amplitude. These high-frequency potentials may begin spontaneously, or may be generated by needle insertion or other mechanical stimulation of muscle such as percussion. Complex repetitive discharges do not change in frequency. They represent the spontaneous discharge of a single myofiber and its surrounding, ephaptically induced myofibers firing in near synchrony. Complex repetitive discharges are most commonly associated with chronic denervation, although they may be seen in myopathies (e.g., myopathy associated with hyperadrenocorticism or hyperkalemic periodic paralysis) (see Figure 27-2, F). Myotonic potentials: So-called myotonic potentials also may be recorded during electromyography. Despite the name, they are not pathognomonic for myotonia, as they also may be seen in patients with radiculopathies and polyneuropathies. Although similar to complex repetitive discharges in many respects, they may be distinguished from complex repetitive discharges by their waxing and waning frequencies (complex repetitive discharges do not change in frequency). This waxing and waning frequency is appreciated on the speaker as phases of increasing amplitudes and frequencies of discharges, followed by decreasing amplitudes and frequencies, which convey the sound of diving propeller-driven airplanes. Consequently, myotonic potentials may also be referred to as “dive bomber” potentials, based on the sounds heard on the speaker. Myotonic potentials are characteristic of myotonia congenita. Peripheral Nerve Conduction Studies: In addition to electromyography, measurement of nerve conduction velocities of both motor nerves and sensory nerves should be performed. Indications for performing these tests are identical to those given previously for electromyography. The definition given by the American Association of electromyography and Electrodiagnosis (AAEE) for nerve conduction studies1 is as follows: “recording and analysis of electric waveforms of biologic origin elicited in response to electric or physiologic stimuli.” In this chapter, only electrical stimulation will be covered. Although techniques may vary between laboratories, the principles remain the same. Ideally, each laboratory should determine normal reference values for individual nerves in each species. Motor Nerve Conduction Velocity Testing: Motor nerve conduction velocity (MNCV) testing involves the placement of a pair of recording electrodes (needle or surface) near a muscle innervated by the nerve of interest, and at least two pairs of stimulating electrodes (usually needle) along the course of that nerve. A ground electrode is used to minimize interference. The active recording electrode is placed subcutaneously over the motor point of the muscle with the reference electrode located several centimeters distally. This placement ensures the optimal configuration of the M-wave (a type of compound muscle action potential) with an initial negative (upward) deflection (Figure 27-3).
Neurodiagnostics
Anatomy of the Motor Unit
Classification of Neuromuscular Disorders
Clinical Signs of Neuromuscular Disorders
Diagnosis of Neuromuscular Disorders
Minimum Database for Neuromuscular Disease
Creatine Kinase
Serum Electrolytes
Blood or Plasma Lactate and Pyruvate
Urine Myoglobin
Thyroid Screening
Acetylcholine Receptor Antibody
Additional Laboratory Testing
Molecular Diagnosis of Inherited Neuromuscular Diseases
Specific Diagnostic Procedures for Neuromuscular Diseases
Electrodiagnostic Testing
< div class='tao-gold-member'>
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree