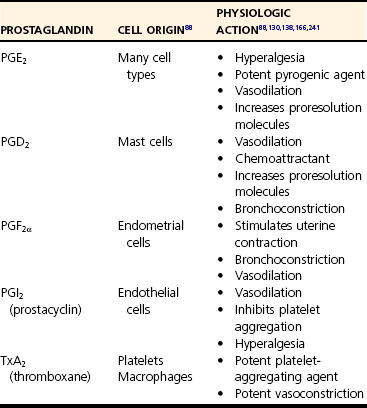
Chapter 1 Upon initial injury, local arterioles may undergo rapid, inconsistent, and transient vasoconstriction, providing some immediate hemostasis. Vasoconstriction is stimulated by vasoactive agents such as catecholamines, serotonin, bradykinin, and prostaglandins that are released from surrounding tissue and by norepinephrine released by the sympathetic nervous system.134 Within minutes, vasodilation and the opening of new capillary beds occur, leading to increased blood flow and local delivery of soluble mediators and inflammatory cells.73 Vasodilation is a consistent component of acute inflammation induced by vascular smooth muscle changes mediated by nitric oxide, histamine, leukotrienes, prostaglandins, and complement factors. Concurrently, lymphatic vessels proliferate to accommodate drainage of edema,4 also serving as an avenue for microbes to reach local lymph nodes. Although these initial responses may be beneficial, an imbalance of mediators may promote widespread vasodilation, systemic hypotension, and even shock, as seen in severe sepsis.217 Vasodilation is followed by an increase in vascular permeability caused by a number of mechanisms. An increase in the number and size of intracellular endothelial gaps in the venules is mediated by histamine and serotonin. These transcytoplasmic channels (vesiculovacuolar organelles) allow transcytosis of plasma products to the site of inflammation.75 Some molecules cannot transverse the transcytoplasmic channels and use other mechanisms. For example, endothelial cell retraction and interendothelial gap formation may occur; these are stimulated by hypoxia, endothelial injury, cytokines, or other inflammatory mediators.75,150,210 Very large plasma molecules and mediators that cannot transverse the endothelial barrier via vesiculovacuolar organelles use interendothelial gaps.75 Vascular permeability is also increased by direct trauma and leukocyte-mediated endothelial damage.150 Increased vascular permeability is accompanied by loss of serum protein. This results in decreased intravascular osmotic pressure, increased blood viscosity, and subsequent increases in interstitial osmotic pressure. These pathophysiologic changes, combined with early increases in hydrostatic pressure, lead to the accumulation of edema fluid in the interstitial space.134,217 Edema facilitates the delivery of beneficial soluble factors (antibodies and acute phase proteins) to the site of inflammation. However, edema, along with alterations in tissue pH and oxygenation, induces localized pain and may precipitate loss of function. Fluid loss leads to increased concentration of erythrocytes in the local vasculature. Hemoconcentration combined with decreased hydrostatic pressure leads to intravascular stasis and congestion. Blood stasis allows increased contact time among erythrocytes, leukocytes, and the vascular endothelium, leading to the next stage of acute inflammation.217 As hemostasis develops, leukocytes begin to marginate along the blood vessel walls. Margination facilitates leukocyte interaction with endothelial cells, primarily in the systemic postcapillary venules and the pulmonary capillaries (Figure 1-1).217 The intimate association promotes weak, transient interactions between the glycoprotein cell adhesion molecules called selectins on endothelial cells and their corresponding ligands on inflammatory leukocytes, for example, the carbohydrate ligand Sialyl-Lewis X.3 The three most commonly described are designated E-, P-, and L-selectins according to their surface expression on endothelial cells, platelets, and leukocytes, respectively. E-selectins are upregulated by proinflammatory cytokines and bind receptor molecules on slow moving, marginated leukocytes.100 Initially, the weak endothelial-leukocyte interactions are susceptible to shear stress from blood flow. Rolling of the leukocyte along the endothelium occurs at a velocity proportionate to blood flow as these weak bonds break and reform.217 As rolling progresses, higher-affinity interactions develop between endothelial cells and leukocytes. Figure 1-1 Neutrophil extravasation. Adherence of leukocytes to the vascular wall results from high-affinity bonds, formed by integrins on the leukocyte surface. Integrins are proteins composed of alpha (CD11a, CD11b, CD11c) and beta (CD18) subunits.217 Proinflammatory mediators increase the expression and binding affinity of these leukocyte integrins.67 Corresponding adhesion molecules on endothelial cells are normally expressed at low levels but are upregulated after exposure to inflammatory mediators, including cytokines, coagulation factors, and histamine.18 As a specific example, the intracellular adhesion molecule-1 (ICAM-1) on endothelial cells interacts with the integrins, lymphocyte function-associated antigen-1 (LFA-1, aka CD11a/CD18), and macrophage antigen-1 (Mac-1, aka CD11b/CD18). Overall, adherence halts the progression of leukocytes within the circulation, making them available for recruitment to the site of inflammation. Genetic deficiencies in adhesion molecules inhibit local leukocyte recruitment during inflammation, leading to recurrent bacterial and fungal infections accompanied by peripheral leukocytosis.146 Once adhered, leukocytes migrate through a process termed diapedesis. Although transcellular migration is possible, diapedesis largely occurs through the interendothelial junctions of postcapillary venules, facilitated by endothelial cell retraction and cell adhesion molecules.192,208 In response to molecular stimuli, adhesion molecules (i.e., ICAM-2) are expressed at concentrated levels near interendothelial cell junctions, interact with leukocyte integrins, and facilitate transmigration.115 In addition, platelet-endothelial cell adhesion molecule (PECAM)-1, which also resides on endothelial cells, facilitates leukocyte transendothelial migration and transmigration through the basement membrane (see Figure 1-1). Once through the endothelial barrier, leukocytes must pierce the basement membrane, a complex process involving both adhesive and proteolytic events facilitated by the leukocyte.237 After reaching the interstitial space, leukocyte migration occurs along chemical gradients of exogenous (bacterial byproducts) and/or endogenous (complement components, chemokines) chemoattractant agents. To achieve this, cells bind to extracellular matrix and secrete degradative enzymes that aid directed movement toward the chemoattractant agents.234 Once at the target site, they perform multiple functions to clean the area and repair tissue. Several aspects of leukocyte recruitment are potential therapeutic targets for controlling harmful inflammation. Currently, antagonists of integrins, selectins, and chemokines are available or in clinical trials. However, similar to the effects of genetic deficiencies in adhesion molecules, targeting leukocyte recruitment has complications, including secondary infections.146 In the majority of mammalian species, neutrophils are the most numerous circulating leukocyte, making them readily available to participate in inflammatory reactions. Typically, neutrophils are the first migratory cells to arrive and take a predominant role in acute inflammation.17,134 Numerous chemoattractants (cytokines, complement components, bacterial products) recruit neutrophils to the site of injury129 and may also activate the neutrophils to perform several functions.17 Neutrophils provide local killing and degradation of bacterial macromolecules via phagocytosis and release of superoxide radicals.35 Within the neutrophil, large azurophil (primary) granules contain microbicidal polypeptides such as myeloperoxidase, defensins, lysosome hydrolases, and neutral proteases. Smaller, specific (secondary) granules contain metalloproteases.66,99 In addition, neutrophils produce several proinflammatory cytokines (interleukin [IL]-1α, IL-β, IL-6, and tumor necrosis factor [TNF]-α) that stimulate further inflammation.17 After exposure to an initial stimulus, neutrophils may become primed through molecular mechanisms that are not completely understood at this time. In response to further stimulation, primed neutrophils exhibit markedly enhanced release of reactive oxygen species and other inflammatory mediators. This priming phenomenon may be a critical component of neutrophil-mediated tissue injury.78,108,112 Once in tissue, the short-lived neutrophil typically undergoes necrosis or apoptosis (programmed cell death).175 Apoptotic neutrophils are phagocytosed and removed by macrophages.156,213 During this process, macrophages begin to release antiinflammatory mediators and decrease production of proinflammatory cytokines, including the neutrophil chemoattractants. Neutrophil populations are largely replaced by macrophages within 24 to 48 hours. However, sepsis and other extreme inflammatory conditions may actually delay neutrophil apoptosis, prolonging the proinflammatory state and promoting tissue damage. Macrophages are integral to the inflammatory response, wound debridement, and tissue repair. Tissue macrophages are resident, sentinel cells responsible for early recognition of inflammatory stimuli and are a major, early source of proinflammatory cytokines.49,143 In addition, when extravasated, circulating monocytes differentiate into macrophages and reside in the provisional fibrin-based extracellular matrix.17,95 Like neutrophils, numerous chemotaxins attract monocytes, including cytokines, fibronectin, elastin, complement factors (C3a, C5a), thrombin, and growth factors (e.g., platelet-derived growth factor [PDGF], transforming growth factor [TGF]-β).17 Activated macrophages debride the affected site through phagocytosis of foreign material, pathogens, and damaged cells. They also secrete collagenases and elastases that dissolve damaged tissue matrix further, facilitating debridement and phagocytosis. Activated macrophages produce proinflammatory cytokines (IL-1β, IL-6, and TNF-α), prostaglandins, and growth factors (PDGF and TGF-α). As the acute inflammatory response resolves, macrophages produce factors that stimulate fibroblasts to produce collagen, aiding in wound repair and healing of the inflamed tissue.17,134 Although lymphocytes evoke attention to acquired immunity, it is now evident that they play a role in early, protective inflammatory responses. In particular, the helper (CD4+) T-cells and the cytotoxic (CD8+) T-cells are major components of cell-mediated immunity. CD4+ cells further differentiate into T-helper-1 (Th-1) and T-helper-2 (Th-2) cells. Under the influence of interferon (IFN)-γ and IL-12, T-cells differentiate into Th-1 cells98 that produce a characteristic cytokine profile, including IFN-γ and IL-2. Th-1 cells exert major influences on macrophages by maximizing the bacterial killing potential of macrophages and stimulating proliferation of cytotoxic T-cells. In sepsis, the early loss of T-cells due to apoptosis contributes to poor outcome that can be reversed by T-cell replacement.113 The IFN-γ produced by Th-1 cells also stimulates immunoglobulin G (IgG)2a production by B-cells.168,217 The Th-2 subset of CD4+ cells primarily functions in helminthic infections and allergic reactions. Exposure to those agents causes prolonged T-cell stimulation and production of IL-4, a promoter of differentiation toward the Th-2 phenotype. Th-2 cells produce IL-4, IL-5, IL-10, and IL-13. Overall this causes suppression of innate macrophage function, an increase in IgG1 and IgE production, and eosinophil activation.168,217 Although the mechanisms are not completely understood, severe tissue trauma can result in a bias toward Th-2 responses. Increased production of IL-4 and IL-10 actually inhibits Th-1 T-cell proliferation178 and may predispose toward infectious complications.62,157 It is evident that T-cells and their products must be balanced for an appropriate inflammatory response. Mast cells are ubiquitously distributed in all organs and degranulate in response to physical trauma, complement factors, microbial products, or neuropeptides. They are the primary source of histamine during acute inflammation. In addition, they release other proinflammatory mediators like serotonin, leukotrienes, prostaglandin metabolites, heparin, and cytokines.7,217 Overall, mast cell degranulation enhances the local inflammatory response. Multicellular organisms use an evolutionarily conserved system to alert the body to infection or cellular damage.22,159 The warning molecules, either exogenous or endogenous, incite intracellular signaling cascades that eventually affect basic cell functions (Figure 1-2). Pathogen-associated molecular patterns are highly conserved microbial molecules, recognized as foreign to the host.22,163 Some common pathogen-associated molecular patterns include lipopolysaccharide, lipoteichoic acid, peptidoglycan, and microbial oligonucleotides. In contrast, danger-associated molecular patterns are endogenous molecules such as fibrinogen, which alert the body to cellular damage initiated by infectious or noninfectious agents.159 High-mobility group B1 was recently recognized as an important danger-associated molecular pattern and an important mediator of late-stage sepsis.22 Under normal conditions, high-mobility group B1 is an intracellular molecule but is released with cellular damage or necrosis. Heat shock proteins (HSPs) are intracellular chaperones that normally regulate proper protein folding.203 They were first identified from cells subjected to thermal stress but are produced in response to other stimuli and are found in the circulation after trauma and surgery.203 HSP60 and HSP70 are produced by activated monocytes and, in turn, stimulate other cells in the innate immune system.200 Pathogen-associated molecular patterns and danger-associated molecular patterns signal the immune system by interacting with cell surface receptors. Figure 1-2 Proinflammatory response to alarm signals. Pattern-recognition receptors are a diverse group expressed on the cell surface, within the intracellular compartment, or in bodily fluids.2,155 The group includes toll-like receptors, scavenger receptors, mannose receptors, C-type lectin-like domain–containing receptors, peptidoglycan recognition receptors, and nucleotide-binding site–leucine-rich repeat receptors (Table 1-1).133 Many pattern-recognition receptors are promiscuous, binding to more than one alarm signal molecule. In addition, a single ligand may bind more than one receptor, ensuring a robust and diverse response. The individual effects of pattern-recognition receptor activation are too numerous to list; however, collectively they initiate the complex cellular responses that result in inflammation. Toll-like receptors are arguably the most important and certainly the most studied of the pattern-recognition receptors. Toll-like receptors are type 1 transmembrane proteins that initiate intracellular signaling cascades, which, in general, activate nuclear factor (NF)-κB and result in altered gene transcription.154 Although more than a dozen toll-like receptors are known, nine are well characterized at this writing (see Table 1-1). TLR4 is a major receptor for lipopolysaccharide (endotoxin). In concert with the receptor CD14 and the soluble mediator lipopolysaccharide binding protein, TLR4 activation increases expression of numerous proinflammatory mediators and modulates the further expression of other toll-like receptors. A bidirectional pathway is present between the nervous and immune systems, facilitated by shared biochemical mediators (cytokines and neuropeptides) interacting with their respective receptors. Therefore, an inflammatory response may alter neural function, and neuronal activity may modify immunologic function.177 With surgically induced trauma, this association is of particular importance because damaged nerves promote inflammation and pain responses. Tachykinins are neuropeptides released from peripheral neurons after stimulation or trauma of sensory nerves. A major tachykinin, substance P, is secreted by inflammatory leukocytes (macrophages, neutrophils, and eosinophils) and by prominent neurons in the respiratory, gastrointestinal, genitourinary, and central nervous systems.30,125,177 Binding to G protein–coupled receptors designated as neurokinin-1 receptors (NK1-Rs), substance P promotes transmission of pain signals.96,109 In addition, substance P binds directly to NK1-R on endothelial cells, initiating local vasodilation and increased venule permeability.30,135 Indirectly, substance P also causes vasodilation and edema by promoting the synthesis of leukotrienes, prostaglandins, and nitric oxide.122,135 In addition to vascular effects, substance P stimulates leukocyte chemotaxis and leukocyte-endothelial cell adhesion, which collectively promotes leukocyte extravasation.96 It also can modulate production of proinflammatory cytokines and enhance degranulation of mast cells, further enhancing acute inflammation.111,119,186,218 In contrast, substance P has proliferative effects on endothelial cells and can stimulate neovascularization,177,253 suggesting a role in resolution of inflammation and tissue repair. However, the overall effects of substance P appear to be proinflammatory; therefore, inhibition of neuropeptides has therapeutic potential.144 The acute vascular response is primarily mediated by two vasoactive substances, histamine and serotonin. Because their active forms are stored within cellular granules, they are among the first mediators released during inflammation. Histamine is produced primarily from mast cells and interacts predominantly with the H1 receptor; however, cells express variable levels of several histamine receptors during different phases of the inflammatory response. The direct vasoactivities of histamine cause arteriolar vasodilation, increased venule permeability, and constriction of large arteries.227 Histamine also enhances vasodilation indirectly through prostaglandin synthesis. In addition, histamine plays a role in allergic inflammation by attracting eosinophils and stimulating nociceptors that induce pruritus.134 Because of a short half-life, the effects of histamine peak within 15 to 20 minutes.150 The rapid onset of histamine activity makes it an elusive therapeutic target with regard to ongoing inflammation. However, receptor antagonists have shown some therapeutic efficacy and are currently under investigation.227 Serotonin (5-hydroxytryptamine) has actions similar to histamine150; however, it is not a major mediator in the acute inflammation response of humans or other nonrodent species.221 In mice and rats, serotonin is released from mast cells, basophils, and some neuroendocrine cells, during platelet aggregation. Therefore, the effects of serotonin on acute inflammation are species dependent and must be considered when physiologic responses across species are compared.183 The term cytokine refers to a very diverse group of small, soluble proteins that act as intercellular messengers during a number of physiologic processes. The group includes tumor necrosis factors, interleukins, transforming growth factors, interferons, colony-stimulating factors, and others (see Figure 1-2). Once referred to as lymphokines, cytokines are actually produced by more than one cell type, and a single cell may produce several different cytokines. Secreted in small concentrations that quickly dissipate, cytokines generally exert their influence locally with autocrine or paracrine cellular effects, but may disseminate and influence cells at distant sites. Cytokines interact with cell surface receptors to initiate intracellular signaling pathways that influence cell functions and the production of more cytokines. Several cytokines may act on the same receptor, and a given cytokine may initiate a response at multiple receptors. This promiscuity in receptor affinity ensures the maintenance of innate immune responses. Cytokines are difficult to categorize because of their diversity. Classifications based on cell of origin, structural homology, molecular mechanisms, receptors, and end functions have been described. None of these systems provide well-demarcated groupings because redundancy and pleiotropism are inherent characteristics of cytokines. Here the cytokines will be grouped by a functional classification. Proinflammatory cytokines (e.g., TNF-α, IL-1β, IL-6) increase the innate immune response, and antiinflammatory cytokines (e.g., IL-10, IL-1ra) attenuate the responses. However, it is important to note that cytokine function may depend upon timing, concentration, and association with other cytokines, highlighting the complexity of immune responses. This discussion will include those cytokines classically regarded as integral to the acute inflammatory response. For comprehensive listings, readers are referred elsewhere.170,200 Although a gross oversimplification, cells exposed to pathogen-associated molecular patterns and danger-associated molecular patterns produce a cascade of cytokines, beginning with TNF-α and IL-1 and followed by IL-6 and the chemotactic cytokines. This leads to production of acute phase proteins, leukocyte influx, and release of other mediators to sustain inflammation (see Figure 1-2). Tumor Necrosis Factor: Tumor necrosis factor (TNF-α) is one of the most studied cytokines. Initially called cachectin, TNF-α was first described for its remarkable antitumor activity and association with cachexia in chronic disease states.128 Activated macrophages are a major source of TNF-α; however, other activated cell types will upregulate TNF-α production.128 TNF-α is produced as a membrane-bound surface protein, cleaved by metalloproteases and released in soluble form. TNF-α interacts with two known receptors, TNFR1 and TNFR2,128 which are found on numerous cell types, suggesting that TNF-α mediates an array of effects. In response to a stimulus, TNF-α concentrations peak quickly.200 Consequently, TNF-α may go undetected in some studies of inflammation, even after major surgical interventions.14 After release, TNF-α initiates production of proinflammatory cytokines (i.e., IL-6), reactive oxygen intermediates,172 chemotaxins, and endothelial adhesion molecules, resulting in invasion of cells at the site of inflammation.43,72 TNF-α causes a wide range of additional effects, including activation of natural killer cells,239 proliferation of cytotoxic T-lymphocytes,123 and T-cell apoptosis.235 These effects are inherently counterregulated in vivo by the release of tumor necrosis factor receptors from the cell surface. These solubilized receptors bind to TNF-α and effectively reduce the cytokine’s activity. Tumor necrosis factor soluble receptors are constitutively released in low numbers but increase in inflammatory conditions such as sepsis. TNF-α release has both beneficial and deleterious consequences.97 Administered experimentally, TNF-α results in classic signs of endotoxic shock, including hypotension, metabolic acidosis, and death.229 Inhibition of TNF-α activity is protective in endotoxic shock.21 Conversely, TNF-α is necessary for protection from mycobacterial infection,81 and blocking its activity increases mortality in septic human patients.80 As a major initiator of inflammation, TNF-α has been directly linked to a number of diseases, leading to interest in TNF-α as a therapeutic target. Although steroids are known to inhibit production of TNF-α, a more focused approach has been applied to specific diseases.200 Commercially available anti-tumor necrosis factor monoclonal antibodies and recombinant tumor necrosis factor soluble receptors have proven efficacious in human patients with Crohn’s disease and rheumatoid arthritis.12,165 It is interesting to note that therapeutic inhibition of TNF-α for rheumatoid arthritis has been associated with recrudescence of pulmonary mycobacterial infection and infectious complications after orthopedic surgeries.23,81,92 In spite of these reports, the success of anticytokine therapy is believed to outweigh the risks. Interleukin-1: The term interleukin-1(IL-1) denotes several cytokines produced by macrophages and other cell types.200 IL-1β is secreted as an inactive proform, which is cleaved by IL-1 converting enzyme, also known as caspase-1.226 However, genetically engineered mice deficient in IL-1 converting enzyme remain responsive to endotoxin, suggesting that redundancy exists between IL-1β and other interleukins. IL-1β complexes with a functional receptor called IL-1RI and a third component, the IL-1 receptor accessory protein (IL-1RAcP), to initiate cellular signaling pathways. Another member of the interleukin -1 family of cytokines, IL-1 receptor antagonist (IL-1ra), serves a counterregulatory function. IL-1ra is actually an antiinflammatory cytokine that competes with IL-1 for receptor sites. Genetically manipulated experimental mice deficient in IL-1ra show exaggerated inflammatory responses, illustrating its importance in IL-1 regulation. IL-1 demonstrates the intricacies of cytokine regulation that may involve several layers of control, including production, processing, receptor availability, and accessory proteins. The proinflammatory functions of IL-1 are similar to those of TNF-α, and these cytokines often work synergistically to further enhance inflammation.172,179 In response to inflammatory stimuli, IL-1 mediates increases in production of proinflammatory cytokines, prostaglandins, and nitric oxide. These changes are manifest in host responses, including hypotension, fever, decreased white blood cell counts, hemorrhage, and pulmonary edema.93,179 Competitive inhibition of the IL-1 receptor improves survival after experimental administration of endotoxin. As with tumor necrosis factor, IL-1 has been implicated in a number of inflammatory diseases, including sepsis, Crohn’s disease, and rheumatoid arthritis. Interleukin-6: Interleukin-6 (IL-6) increases in virtually all inflammatory conditions. IL-6 is produced by macrophages, T-cells, epithelial cells, and enterocytes. It plays a pivotal role in initiating hepatic synthesis of the acute phase proteins185,200 and influences the proliferation of lymphocytes. In addition, IL-6 has a contradictory role in initiating compensatory responses by inducing antiinflammatory responses and downregulating proinflammatory cytokine production.5,243 In inflammatory states, plasma IL-6 increases proportionately with the duration216 and severity of the condition. After surgical trauma, plasma levels are higher with invasive procedures58,86 as compared with laparoscopy.126,232 IL-6 levels have been used to predict postoperative infection161 and mortality associated with sepsis.197 IL-6 may also predict the possibility of recurrent abdominal adhesions.50 Consequently, IL-6 is considered to be not only a mediator but also a diagnostic and prognostic biomarker of inflammation. Chemokines: In acute inflammation, chemokines peak shortly after TNF-α and IL-1, along with IL-6. Chemokines are the chemotactic cytokines responsible for attraction of cells across a concentration gradient. More than 40 known chemokines are secreted by macrophages and endothelial and other cell types to recruit cells during embryonic development, wound healing, angiogenesis, and inflammatory responses.200 As with all cytokines, redundancy in cell specificity, receptor affinity, and function is noted among the chemokines.13 Therefore, chemokines are categorized into families according to structural placement of conserved cysteine residues (e.g., CXC chemokines have one amino acid separating two cysteine residues). Of the four chemokine families, CXC and CC chemokines contain members most actively involved in the proinflammatory response to trauma or infection. Within the CXC family, a subgroup carries an ELR moiety (glutamine-leucine-arginine), conferring the ability to attract neutrophils, while an ELR negative subgroup attracts mononuclear cells. Interleukin-8 (IL-8) is the archetypical neutrophil chemoattractant in the majority of mammals and, under the most recent nomenclature, is referred to as CXCL8.1 It is noteworthy that rodents commonly used in inflammation research do not express IL-8/CXCL8 but have several functional counterparts. IL-8/CXCL8 attracts neutrophils, upregulates surface expression of adhesion molecules, triggers degranulation of proteases, and promotes production of other inflammatory mediators. As the inflammatory response continues, additional chemokines, such as monocyte chemoattractant protein-1 (MCP-1/CCL2) and macrophage inflammatory protein (MIP-1α/CCL3), participate in the recruitment of monocytes, promoting a transition from active to chronic phases of inflammation. Over time, cellular recruitment slows as chemokines are degraded by enzymes and further production slows. Interleukin-10: Although antiinflammatory cytokines are numerous, interleukin-10 (IL-10) is the archetype. IL-10 is produced primarily by CD4+ Th-2 cells, monocytes, and B-cells.180 It depresses the production of several proinflammatory cytokines and chemokines, including TNF-α, IL-1, IL-6, and IL-8, by inhibiting translocation of nuclear factor κB (NF-κB) and promoting degradation of messenger RNAs.54,180 IL-10 downregulates Th-1 cytokines, which are protective during microbial infection,8 and plays a role in limiting inflammatory responses to normal gut-associated bacteria.180 In addition, IL-10 promotes shedding of tumor necrosis factor receptors into the systemic circulation.120 It also inhibits antigen presentation by macrophages and dendritic cells.180 In a balanced immune response, IL-10 levels would be low during acute phase inflammation and would increase over time. IL-10 deficiencies have been reported in chronic inflammatory, autoimmune diseases and after transplantation surgeries, which may contribute to poor outcomes.180 Conversely, exogenous IL-10 has been used to reduce intestinal inflammation in human patients with Crohn’s disease.8 However, excess IL-10 can increase susceptibility to microbial infection and may influence survival.136 This illustrates that a fine balance of cytokines is necessary to ensure appropriate inflammatory responses. Eicosanoids are lipid mediators that are rapidly synthesized de novo from cell membrane phospholipids and exert their effects locally Their precursor is the fatty acid, arachidonic acid, which is stored in the cell membranes of endothelial cells, leukocytes, and other cells. Arachidonic acid is released by activated phospholipase A2 and is rapidly metabolized by the cyclooxygenase or lipoxygenase pathway (Figure 1-3).29,107 Glucocorticoids suppress inflammation by decreasing phospholipase A2 expression, resulting in decreased production of arachidonic acid. Also, glucocorticoids upregulate genes encoding antiinflammatory proteins that inhibit arachidonic acid release from phospholipids.36,59 Figure 1-3 The arachidonic acid pathway. Prostaglandins: Prostaglandins are produced in the cyclooxygenase pathway, where arachidonic acid metabolism is catalyzed by the enzymes cyclooxygenase (COX)-1 and COX-2. COX-1 is a constitutively expressed enzyme involved in homeostasis and present in the majority of mature cells. Expression of COX-2 is induced by trauma, growth factors, proinflammatory cytokines, and other mediators.88,195 Prostaglandins mediate many inflammatory responses primarily through G protein–coupled receptors on a number of cell types (Table 1-2).29,166 Prostaglandins are chemotactic agents that cause recruitment of leukocytes. Prostaglandins also induce vasodilation and contribute to the pathogenesis of pain and fever during inflammation.241 Aspirin and nonsteroidal antiinflammatory drugs (e.g., carprofen, indomethacin) inhibit the cyclooxygenase enzymes. Selective inhibition of the inducible COX-2 while sparing the constitutively produced COX-1 has received a great deal of attention. It was initially believed that inhibition of COX-1 caused gastric ulceration and thus should be spared. However, in clinical trials in humans, inhibition of COX-2 alone increased the risk of cardiovascular and cerebrovascular events,195 probably through the as yet ill-defined role of COX-2 in vascular homeostasis.103,124,228 In addition, COX-2 may actually help resolve acute inflammation and heal gastric ulcers. Thus, the use of selective COX-2 inhibitors for treating chronic inflammation has gone out of favor in human medicine.27,195 However, no compelling evidence suggests that dogs develop cardiovascular events with COX-2 inhibitor use. This, combined with the decreased incidence of gastric ulceration, makes COX-2 selective agents a good option in dogs.54 Leukotrienes: Leukotrienes are produced in the lipoxygenase pathway, where lipoxygenase enzymes act on arachidonic acid to form the major types of leukotrienes, LTB4, and the peptidoleukotrienes (LTC4, LTD4, and LTE4), which are proinflammatory modulators of leukocyte trafficking and blood flow (see Figure 1-3). Leukotrienes are primarily secreted by leukocytes but are also produced by platelets and endothelial cells.29,82 LTB4 is a potent chemotactic agent and an activator of neutrophils, potentiating their extravasation, degranulation, and production of free radicals.189 The autocrine activity of LTB4 on leukocytes results in cyclic production of leukotrienes during acute inflammation. In addition, the peptidoleukotrienes provoke vasoconstriction, bronchoconstriction, and increased venule permeability.107,189 In general, leukotrienes are more potently vasoactive than histamine.106 Consequently, agents that block leukotriene production or antagonize leukotriene receptors have been used to treat both inflammation and airway responsiveness associated with asthma.107
Inflammatory Response
Acute Inflammation
The Acute Vascular Response
Permeability
Stasis
Leukocyte Extravasation
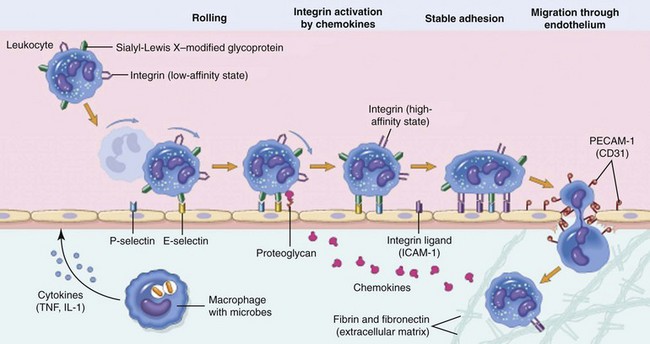
Leukocyte extravasation is a multistep process orchestrated by both hemostatic and cell–cell interactions. Margination and rolling of leukocytes along the vascular endothelium are mediated through interactions between endothelial selectins with their corresponding leukocyte ligands. Chemokines stimulate increased expression and enhanced binding affinity of leukocyte integrins, leading to firm adherence to endothelial cell integrins (e.g., intracellular adhesion molecule [ICAM]-1). Leukocyte diapedesis is facilitated by the adhesion molecule, platelet–endothelial cell adhesion molecule (PECAM)-1, and leukocytes follow chemokine gradients to the site of injury. IL-1, Interleukin-1; TNF, tumor necrosis factor. (From Kumar V, Abbas A, Fausto N, Aster J: Robbins and Cotran pathologic basis of disease, ed 8, Philadelphia, 2009, Saunders/Elsevier.)
Cellular Components
Neutrophils
Macrophages
Lymphocytes
Mast Cells
Inflammatory Stimuli
Alarm Signals: Pathogen-Associated Molecular Patterns and Danger-Associated Molecular Patterns
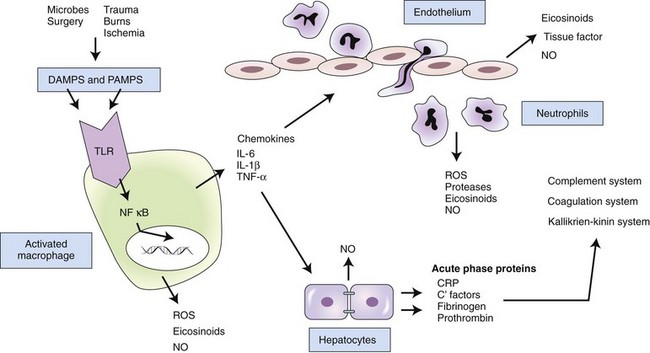
In response to pathogen-associated molecular patterns (PAMPS) or danger-associated molecular patterns (DAMPS), toll-like receptors (TLRs) on the surface of resident macrophages induce various molecular signaling pathways. Many of these pathways lead to the translocation of nuclear factor kappa B (NFκB) into the cell nucleus, where it acts as a transcription factor, regulating the production of proinflammatory cytokines. The cytokines act directly and indirectly on a number of cell types. Interleukin (IL)-6 induces hepatic production of acute phase proteins, which in turn influence a number of inflammatory systems. Chemokines induce recruitment of inflammatory cells, which produce additional mediators. If the process is not properly balanced by antiinflammatory responses, tissue damage and systemic inflammation may result in serious consequences. CRP, C-reactive protein; NO, nitric oxide; ROS, reactive oxygen species.
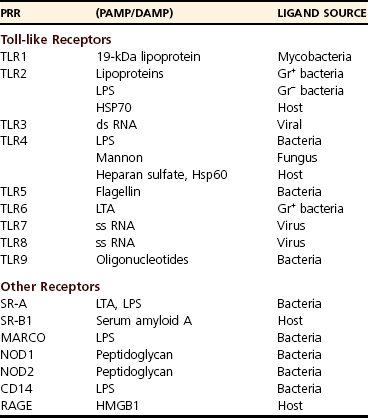
Pattern-Recognition Receptors
Toll-like Receptors
Neurogenic Inflammation
Tachykinins
Mediators of Inflammation
Cytokines
Proinflammatory Cytokines
Antiinflammatory Cytokines
Lipid/Cell Membrane–Derived Mediators
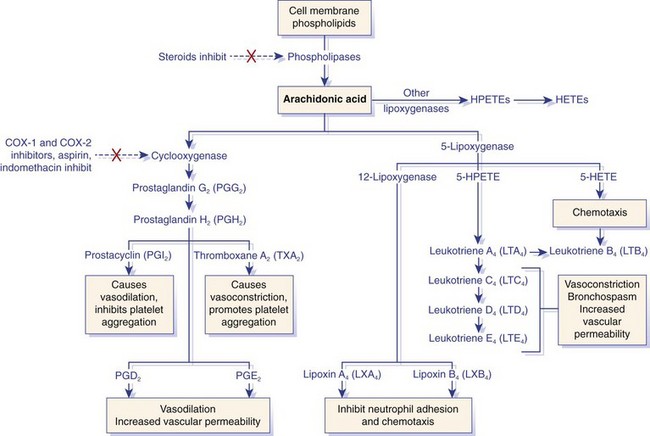
Arachidonic acid is metabolized by the cyclooxygenase or lipoxygenase pathway to produce prostaglandins or leukotrienes and proresolution lipoxins, respectively. The inhibitory effects of several drugs on specific enzymes are denoted by a red X. COX, Cyclooxygenase; HETE, hydroxyeicosatetraenoic acid; HPETE, hydroperoxyeicosatetraenoic acid. (From Kumar V, Abbas A, Fausto N, Aster J: Robbins and Cotran pathologic basis of disease, ed 8, Philadelphia, 2009, Saunders/Elsevier.)
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Inflammatory Response
Only gold members can continue reading. Log In or Register to continue
