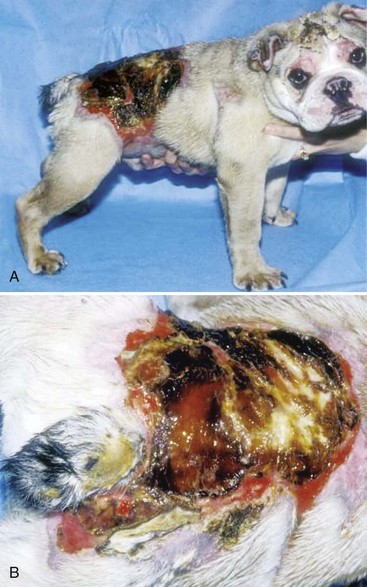
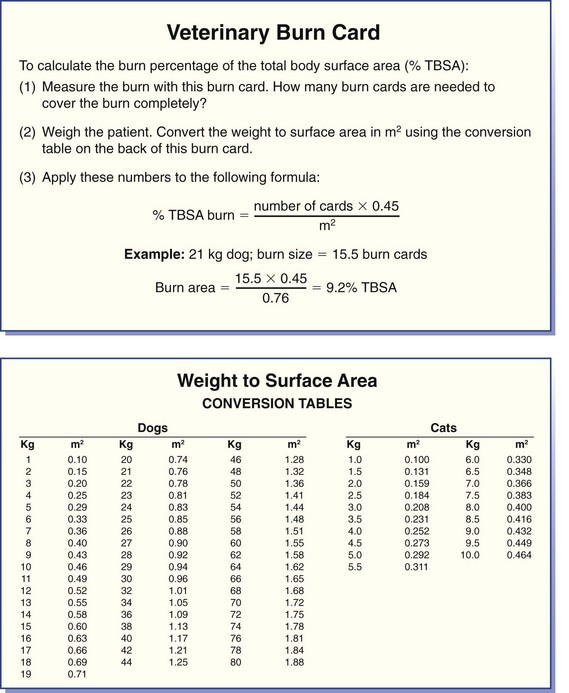
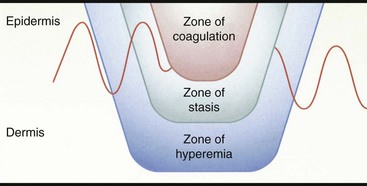
Chapter 81 Burn injuries in small animals can present in a wide clinical range, from mild burns that heal rapidly with no treatment to large and deep burns, which are among the most difficult and challenging wounds to treat in dogs and cats and carry a serious to grave prognosis. Successful management of the latter types of cases hinges on a thorough grasp of the pathophysiology of the local burn wound and the systemic response to burn injury. Burns are a common injury in humans, with an estimated 1.25 million cases per year in the United States, resulting in 70,000 to 80,000 hospital cases.60 Because serious burns in companion animals are relatively uncommon, however, most treatment recommendations for animals come from the knowledge garnered from clinical studies in human burn victims. In turn, principles of burn treatment applied to humans are largely based on animal experimentation, with canine models frequently used. This direct connection helps validate the application of much of the human case data to the care of small animal burn patients as long as proper consideration is given to anatomic and physiologic differences between humans and animals. All burns, regardless of their cause, occur when tissue is exposed to an energy flux of sufficient rate or magnitude to exceed tissue capacity to dissipate the energy, resulting in the creation of the characteristic burn lesion. The source of the energy determines the classification of the burn into one of four basic etiologic categories:95,111 (1) tissue exposure to temperature extremes (either high or low) sufficient to cause cellular damage is the cause of thermal burns; (2) exposure to chemicals that cause tissue necrosis via chemical reactivity (or secondary thermal effects) results in chemical burns; (3) electrical burns occur when an electrical current of sufficient energy passes through the patient, causing cellular necrosis along its path; and (4) radiation burns are caused by exposure to ionizing radiation at levels that cause acute cell death, an injury that is most commonly seen from exposure to solar radiation or as a side effect of radiation therapy for neoplastic disease. Accurate determination of the cause of a burn and knowledge of its pathogenesis is an important determinant of both the initial care of the burn wound and the prognostication of outcome. Thermal burns include injuries secondary to exposure to either excessive heat (hyperthermic burns) or excessive cold; therefore, frostbite is technically a “thermal” burn. Common use of the term thermal burn is restricted to hyperthermic injury, however, and this convention will be followed in this chapter. Thermal burns may be classified according to the heat source, that is, (1) a flame or fire, (2) scalds from hot liquids or gases, and (3) burns from direct contact with a hot object.46 Regardless of the heat source, thermal burns occur when heat is transferred to the tissues in one of three ways: via conduction (direct contact with a hot object), convection (airborne heat transfer such as the superheated air in a house fire), or radiation (electromagnetic energy interacts with the body and is converted to heat). Depth of Tissue Destruction: The severity of burn injury is measured by the depth of tissue destruction. Two schemes of classification are in widespread use. The first recognizes five degrees of burn injury based on depth. First-degree burns are the most superficial; only the epidermis is affected, and erythema is the only grossly observed initial sign. Healing is evidenced by the premature sloughing of damaged epidermis; no blistering or open wounds are seen and, because the dermis is intact, there is no scarring. However, despite the limited depth of damage, first-degree burns are painful because of nociceptor stimulation at the time of injury and during the subsequent inflammatory response. Second-degree burns extend into the underlying dermis, with resultant fluid exudation and blistering. Blistering is rare in dogs and cats but common in humans because of differences in dermal histology. Third-degree burns extend completely through the dermis to the underlying subcutaneous tissue. Some authors recognize two additional categories: fourth-degree burns that extend to underlying muscle or fascia and fifth-degree burns with extension to bone.111 A simplified description of burn severity has two classifications: partial-thickness burns (first and second degree) and full-thickness burns (third degree and higher; Figure 81-1). This is a practical scheme for most clinical applications because the loss of the dermis is a common factor in serious burns that dictates many treatment decisions. The degree of tissue damage is directly proportional to the temperature and duration of exposure.8 At 40° to 44° C, failure of the cell membrane sodium pump occurs; epidermal necrosis (partial-thickness burn) occurs if the skin temperature reaches 60° C for one second;62 skin temperatures above 70° C result in full-thickness burns in less than 1 second.76 Surface Area: Burns are also classified by the area of affected skin. Several methods have been developed to estimate the extent of burns as percentage of total body surface area. A simple method, developed for human patients, is Wallace’s “rule of nines,” which divides the body into regions that are multiples of 9% of total body surface area. This method has been adapted for use in veterinary patients. In animals, the head and neck are counted as one “nine” or 9%. Each forelimb is also 9%, each hindlimb two “nines” or 18%, and the dorsal and ventral halves of the trunk also 18% each.27 In human medicine, it is widely recognized that the rule of nines has limitations: body shape (adult versus infant) and body condition (normal versus obese) have major effects on the accuracy of the system.63 Given the equal or even greater variability of shapes and surface areas in the animal world, it is quite possible that the rule of nines may be subject to similar or perhaps even greater errors of estimation in veterinary applications. Another total body surface area estimation tool has been developed that may be more consistent, the “Resuscitation Burn Card,” which is based on the fact that an ordinary plastic credit card is standardized to 8.5 × 5.3 cm or 45 cm2. The size of the card is compared with the burn to determine the total burn area in cm2 for the numerator. The denominator is obtained from a standardized body weight to surface area chart such as is used in dosing of antineoplastic chemotherapeutic drugs, and the percentage of total body surface area is calculated (Figure 81-2).69 Local Response to Thermal Burn Injury The severity of a burn injury radiates outward from the greatest point of energy delivery to the tissue; three regions of functional significance are recognized (Figure 81-3). The inner area is called the zone of coagulation (also called the zone of necrosis or zone of destruction); no viable tissue remains in this zone. The zone of stasis is the next area, so named because of its reduced perfusion. Decreased perfusion occurs because of reduction in deformability of erythrocytes secondary to heat-damaged proteins in the cell membranes and reduction of vascular luminal diameter from increased interstitial pressure secondary to increased capillary permeability. Increased capillary permeability depends on the severity of the burn: in a canine model of scald burns, local lymph flow and protein content increased proportionally with scald temperature.25 Tissues in the zone of stasis are vulnerable; whereas further insult leads to necrosis, effective therapy can restore perfusion and maintain viability. The outermost area is the zone of hyperemia, which is the primary area of the inflammatory response to the burn. Tissues in the zone of hyperemia are viable and heal if no further injury is sustained.49 The local inflammatory response to burn injury is characterized by vasodilation, increased capillary permeability, edema, and influx of inflammatory cells.33 Damaged tissue and inflammatory cells resident at the time of injury are the primary sources of chemokines that initiate the inflammatory response, including endotoxin,41 prostaglandin E2,40 histamine, and activated complement.28 Local perfusion is upregulated immediately after burns because of arteriolar vasodilation; this occurs in response to postganglionic autonomic stimulation113 and upregulation of nitric oxide synthesis within the area of thermal injury92 and surrounding unburned skin.85 Nitric oxide acts as a potent vasodilator in burns via direct effects on vascular smooth muscle and indirectly by stimulation of release of other vasodilatory cytokines, such as substance P.133 Along with increased perfusion, capillary permeability also increases in the immediate postburn period; it is also mediated by the inflammatory cascade and is proportional to burn severity.25 The combination of increased perfusion and capillary permeability leads to postburn edema. Burn wounds often heal slowly compared with normal surgical wounds. One apparent reason is the markedly lower concentrations of wound healing cytokines found in burn wounds. Wound fluid from burns has been shown to have less than 5% of normal levels of fibroblast growth factor-2 (FGF-2) and none of the capillary endothelial chemotactic and proliferative activity seen in normal surgical wounds.83 Pulmonary System: Smoke Inhalation Smoke inhalation is a common injury that occurs concurrently with burns, particularly when the victim is indoors. Smoke inhalation produces a complex and interrelated array of pathophysiologic changes in many organ systems, principally as a result of two processes: (1) the local and systemic inflammatory response to smoke exposure and (2) failure of oxygen delivery at the tissue level. Smoke exposure consists of two components: thermal and toxic. The former component is usually limited to the upper airways because of rapid cooling of hot air after it has been inhaled. Hot air entering the larynx at over 500° F is cooled to 120° F before it reaches the lungs because of the low heat-carrying capacity of dry air and rapid conduction of heat via the tracheal mucosal blood supply. Therefore, the majority of the injury from smoke inhalation is caused by the toxicity of smoke. Smoke from house fires contains more than 250 toxic substances.86 These include carbon monoxide from incomplete combustion of carbon; hydrogen cyanide from combustion of nitrogen-containing products (e.g., nylon, formica, melamine, wool); and inorganic acids (hydrochloride, hydrogen fluoride, and hydrobromic acid) from combustion of polyvinyl chloride, Teflon, neoprene, and plastics. Carbon monoxide exerts toxic effects via three mechanisms: (1) preferentially binding to hemoglobin (forming carboxyhemoglobin), thereby reducing its oxygen-carrying capacity; (2) carboxyhemoglobin formation, resulting in a leftward shift of the oxyhemoglobin dissociation curve and reducing oxygen delivery to the tissues; and (3) binding of carbon monoxide with myoglobin to reduce oxygen availability to muscle, particularly cardiac and skeletal. Hydrogen cyanide binds with mitochondrial cytochrome oxidase, disrupting the electron transport chain and preventing cellular respiration. Hydrochloride and the other diatomic halide acids are all intensely irritating to respiratory mucous membranes, producing laryngospasm and bronchospasm at concentrations found in smoke.3 Smoke inhalation also causes a dose-dependent injury to tracheobronchial epithelium and lung parenchyma via free hydroxyl and carbon radicals contained in the smoke and via the accompanying neutrophilic infiltration.86 The lung’s response to smoke inhalation consists of a number of pathophysiologic changes, including increased pulmonary vascular permeability80 and venoconstriction,81 which cause rapid accumulation of fluid, mucus, and neutrophils within the alveoli and airways, hallmarks of pulmonary edema.18 Other pathophysiologic changes include atelectasis,38 decreased alveolar ventilation,124 deactivation of pulmonary surfactant,82 and decreased lung compliance.71 Together these changes result in the development of acute respiratory distress syndrome (ARDS).23 Among the numerous effectors of pulmonary edema and the other pathophysiologic changes in smoke inhalation injury are activated neutrophils, eicosanoids, cytokines, nitric oxide, free radicals, and neurotransmitters such as substance P.77,127 These derive three main sources: smoke-damaged lungs; burn-injured tissues; and the gastrointestinal tract, from which cytokines travel to the lungs via lymphatics. The eicosanoid thromboxane A2, synthesized in pulmonary macrophages and released in response to smoke inhalation, causes an increase in pulmonary transvascular flux and vascular resistance secondary to marked pulmonary venoconstriction.81 Gastrointestinal-derived cytokines act synergistically with endotoxin and translocated gut bacteria, causing increased pulmonary vascular permeability and apoptosis of alveolar lining.67 In addition to local vascular leakage, systemic extravasation of fluid from the vascular space is one of the early pathophysiologic changes seen with serious burns (>25% total body surface area).91 Within 10 minutes after a burn, systemic vascular permeability to fluid and albumin increases because of myosin-mediated contraction of vascular endothelial cells and direct damage to endothelial cells.22 These effects are mediated by complement activation, histamine, and oxygen free radicals from the burn site.28 Systemic extravasation rapidly overwhelms the lymphatic system, and generalized edema and hypovolemia ensue as protein-rich fluid accumulates in the interstitial space. This process usually peaks in the first 12 hours after burn and can be quite marked; in a canine model, 20% total body surface area burns caused a 28% loss of plasma volume within 6 hours postburn. Evaporation is another significant source of fluid loss leading to hypovolemia: direct fluid losses from a full-thickness burn wound are three to 20 times greater than intact skin,9,35 and even a partial-thickness burn can increase evaporation by 33% compared with intact skin.16 The combination of extravasation and evaporation creates profound hypovolemia within hours after a serious burn. In addition, erythrocyte deformability is decreased, and aggregation is increased secondary to peroxidation of membrane lipids.5,6 The combination of hypovolemia and decreased erythrocyte deformability causes hyperviscosity and degrades the rheologic (flow) characteristics of blood. The condition is exacerbated by systemic vasoconstriction, which is proportional to burn total body surface area101,106 and is mediated by sympathetic response to baroreceptor and nociceptor afferents.13,100 The combined effects of hypovolemia, hyperviscosity, and vasoconstriction cause hypoperfusion and thrombosis, leading to tissue hypoxia and metabolic acidosis.13,55,65 The systemic inflammatory response to large burns also produces direct negative effects on myocardial function. Significantly decreased left ventricular contractility was observed after 50% total body surface area burns in dogs112 caused by an increase in cardiac myocyte intracytoplasmic Ca2+.128,130 Intracytoplasmic Ca2+ increases as a result of a combination of mechanisms. One mechanism is oxidative injury to the sarcoplasmic reticulum,21 which induces Ca2+ leakage into the cytoplasm and reduces sarcoplasmic reticulum reuptake of Ca2+.21,78 Burn injury also causes cytosolic sodium accumulation in cardiac myocytes;129 this has been hypothesized to enhance sodium–calcium ion exchange, thus further contributing to cytosolic Ca2+ accumulation.130 These ionic derangements occur secondarily to the burn inflammatory response and have been linked to gastrointestinal translocation of bacteria, endotoxin, and cytokines and burn systemic inflammatory response syndrome (SIRS).48,132 Myocardial damage and decreased cardiac output123 are also seen as secondary effects of carbon monoxide intoxication from smoke inhalation. In one canine model of smoke inhalation injury, increased carboxyhemoglobin concentration was associated with decreased cardiomyocyte adenosine triphosphate production and enzymatic and histopathologic evidence of cardiomyocyte necrosis.90 The gastrointestinal system is profoundly affected by large burns and is, in turn, a major effector organ for burn SIRS and multiple organ failure. After a severe burn, gastrointestinal barrier function is compromised; subsequent translocation of gut bacteria, endotoxin, and cytokines leads to septic shock.36,54,66 Burn injury increases the apoptotic rate of gut mucosal cells with no corresponding increase in mucosal proliferation; this may be an important factor in postburn loss of gastrointestinal mucosal integrity.52 Postburn gastrointestinal motility is also impaired; this is mediated via an increased expression of inducible nitric oxide synthase by neurons of the myenteric plexus.30 The liver is also affected by burn injury: burns cause increased oxidative stress in hepatocytes.84 Hepatocyte turnover is also increased, shown by increased rates of apoptosis and proliferation.53 Increased circulating catecholamine concentrations promote development of hepatic lipidosis.19 Liver synthetic function is also affected: upregulation of certain products such as acute phase proteins51 and downregulation of albumin production107 has been noted by 3 days after burn injury. The incidence of acute renal failure in human patients with severe burns ranges from 1.3% to 38%, with polyuric acute renal failure predominating; acute renal failure is associated with a high mortality rate between 73% and 100%.97 Burn severity (as percentage of total body surface area) is an independent predictor of the likelihood of acute renal failure and associated mortality.57 A number of contributory factors have been identified, including hypotension, hypoalbuminemia, hemoglobinemia, myoglobinemia,57 sepsis,47,57 and use of nephrotoxic antibiotics.47 In addition, reduced cardiac output and systemic vasoconstriction contribute to acute renal failure; increased concentrations of stress hormones (catecholamines, vasopressin, angiotensin, and aldosterone) have been implicated in the mechanism.1 Atrial natriuretic peptide, which is also increased in postburn patients, may play a protective role by increasing renal blood flow and urine output. Dogs receiving a constant norepinephrine infusion responded to exogenous atrial natriuretic peptide administration with improvement in renal and hemodynamic parameters.1 Acute renal failure may also develop as a late complication of burn injury. A multifactorial cause is suspected, including delayed effects of renal ischemia, sepsis, nephrotoxic effects of antibiotics or other drugs, and glomerular accumulation of hemoglobin and myoglobin and other debris from necrotic cells.47,95 Burn injury produces an immediate and long-lasting reduction in circulating erythrocyte numbers (“burn anemia”), which is the result of a combination of increased red blood cell (RBC) loss and decreased erythropoiesis. Up to 10% of the circulating RBC mass may be trapped and destroyed in a large burn.64 This only accounts for a portion of the total RBC loss: human burn patients average 12% loss of RBC mass within 6 hours of a large (15% to 40% total body surface area) burn and may lose up to 18% of their RBC mass within 24 hours.118 Within 1 hour of a burn injury, the concentration of free hemoglobin in the plasma increases, signaling intravascular hemolysis, which occurs because of membrane damage that increases erythrocyte fragility and decreases erythrocyte deformability. In a rat model, complement activation led to free radical production by neutrophils, with resultant intravascular hemolysis; the effect peaked at 15 minutes after burn injury but continued at elevated levels for more than 2 hours.43 Decreased erythrocyte deformability is also caused by lipid peroxidation of the RBC membrane, associated with a decline in protective levels of the antioxidants glutathione and α-tocopherol.5,7 The loss of RBC mass stimulates an appropriate increase in erythropoietin release from the kidneys; nonetheless, erythropoiesis remains depressed.4,122 Reduced erythropoiesis appears to be caused by decreased iron availability by an inhibitory protein of 50 kilodaltons (kD) molecular weight122 and as a secondary effect of decreased iron availability.4 Administration of supplemental exogenous erythropoietin helps restore RBC mass after a burn.26,102 Burn injury produces significant negative effects on lymphocyte production and function. For example, lymphoid apoptosis is upregulated after burn injury and appears to be linked to the upregulation of tumor necrosis factor-alpha (TNF-α).17 Burn injury also causes increased susceptibility to sepsis because of inhibition of chemotactic cytokine production by T-cells. The sympathetic nervous system appears to mediate this effect: chemical sympathectomy normalized cytokine production and improved resistance to sepsis in an in vivo mouse model. In an in vitro model, reduced cytokine production was also noted in T-cells from burned mice or norepinephrine-treated T-cells from normal mice.114 Burn injury also alters the function of macrophages and neutrophils. Burns stimulate macrophages to express a hyperinflammatory phenotype, with overproduction of tumor necrosis factors, interleukins, nitric oxide, and eicosanoids. This hyperinflammatory macrophage phenotype appears to play a role in the increased postburn susceptibility to sepsis99 and to be modulated by T-cells: induction to a hyperinflammatory phenotype was partially blocked in T-cell–deficient mice, resulting in improved survival to a septic challenge.98 Neutrophils also become activated to a hyperinflammatory phenotype after burn injury with marked upregulation of superoxide production; increases in intracytosolic Ca2+ and protein kinase C appear to be responsible.96 At the same time, neutrophil migration or chemotaxis is depressed, and adhesion to vascular endothelium is increased. The combination of these factors leads to increased potential for neutrophil-mediated intravascular damage after burn injury.121 All burns cause intense pain that may seem to be out of proportion to the apparent severity of tissue damage. This is a result of the local burn inflammatory response in injured tissue that retains viable cutaneous nociceptors, which send afferent input along A δ and C fibers.109 Cellular damage and local inflammation also stimulate release of chemical pain mediators such as prostaglandins and kinins; these further sensitize local nociceptors into a hyperalgesic state.29 The resultant intense pain stimulates a massive sympathetic discharge, which promotes the cardiovascular events of burn shock. Even after the initial burn shock is resolved, tissue damage and mechanical stimulation of the wound create a chronic painful state that fuels an ongoing release of catecholamines at a lower level. This chronic catecholamine release is a mediator of many of the metabolic and organic responses to burns.87 Peripheral nociceptors also modulate the initial local inflammatory response to burn injury via effects on vasomotor tone and inflammatory cell chemotaxis. In a scalded rat hindpaw model, substance P and calcitonin gene–related peptide released from peripheral sensory neurons induced vasodilation in the injured tissue. In addition, neuropeptides have been demonstrated to induce chemotaxis and activation of neutrophils, eosinophils, mast cells, and monocytes after tissue injury.56 Substance P also acts a mediator of pulmonary injury after smoke inhalation: pulmonary release of substance P has been demonstrated to be one of the effectors of the increased vascular permeability and pulmonary edema that ultimately lead to ARDS.127 Burn injury produces profound changes in energy and protein metabolism mediated by the interaction of local effects (e.g., oxidative stress131 and release of proinflammatory cytokines such as TNF-α, interleukin-6 [IL-6], and IL-8)103 and systemic effects from increased release of catabolic hormones (primarily cortisol and catecholamines).125,131 In the immediate postburn period, the body enters a period of hypometabolism (the “ebb phase,” which occurs during acute phase burn shock) followed by a hypermetabolic “flow phase.” Basal energy expenditure during the flow phase increases by more than 100% compared with preburn,12,125 driven by increased heat loss and an increase in the hypothalamic set point. Loss of the barrier function of skin causes a large increase in evaporation, with concomitant heat loss as the heat of evaporation. The hypothalamic “set point” is increased by 1° to 2° C in response to the release of inflammatory cytokines and eicosanoids. Nonproductive metabolic work produces the required heat at significant energy cost to the patient. For example, metabolic studies in human burn patients report a 450% increase in triglyceride–fatty acid cycling and a 250% increase in glycolytic–gluconeogenic cycling.45 Utilization of proteins and carbohydrates is altered in the postburn state. Amino acids are used for energy production, resulting in a loss of lean body mass as protein is catabolized. In normal prolonged fasting, adipose lipids supply 90% of the basal energy requirement with lean body mass supplying only 5% to 8% of energy needs. This is compared with the postburn state, in which adipose supplies about half of the energy requirement and protein catabolism accounts for approximately 30%.20 Upregulation of hepatic gluconeogenesis and relative insulin resistance results in persistent hyperglycemic and catabolic states marked by glucose intolerance and hyperinsulinemia. Although most peripheral tissues (e.g., muscle, fat) experience reduced glucose uptake, the burn wound has an increased rate of glucose uptake because of its high rate of anaerobic glycolysis by inflammatory and endothelial cells and fibroblasts.45 This condition, so-called “burn diabetes,” exists in acute and chronic stages of burn healing and is a serious systemic complication of burn injury because increasing loss of lean body mass is linked to degradation of prognosis.20 If possible, first aid care should be given to the burned small animal patient before presentation to a veterinarian. The most well-documented first aid therapy is the simple application of cool to cold running water (2° to 15° C or ~35° to 60° F) directly to the burn wound. In addition to the obvious analgesia provided by cooling, studies have shown beneficial effects on burn wound healing as well. In a porcine model of deep partial-thickness burns, immediate application of 15° C water for 20 minutes resulted in improved healing as measured by increased wound epithelialization and reduced scar size at 6 weeks postburn.11 Although immediate cooling gave the best results, measurable benefit can still be seen with first aid delayed for up to 3 hours postburn. The optimum temperature appears to be cool water at 15° C; cold water at 2° C brought about a more rapid reduction in subdermal temperature and slightly more rapid contraction but no improvement in epithelialization. Cooling with ice produced results similar to untreated control subjects and is contraindicated.94 Numerous other topical treatments have been advocated for burn first aid; most claim to have analgesic properties, and some also claim improved healing, mostly without documentation of efficacy. In one comparison of the topical application of aloe vera, tea tree oil, and saliva to burns, none of the treatments produced improvement in healing or reduced postburn wound infection compared with untreated control subjects.15 Based on the experimental data, at the present time, the use of these and other alternative therapies for burn first aid is not recommended.2 After it has been cooled, the burn should be covered with a sterile, occlusive, nonadherent dressing before transporting the patient for definitive care. Covering the burn reduces pain and protects it from contamination and further trauma. The Brooke formula developed by Artz at the Army Burn Center includes crystalloids and colloids because use of colloids in resuscitation was believed to be important at the time. The modified Brooke formula proposed by Pruitt in 1971 is perhaps the second most widely recognized burn resuscitation formula after the Parkland. The modified Brooke formula prescribes lactated Ringer’s solution at 2 mL/kg/%burn. One half of this dose is given during the first 8 hours, and the second half is given over the next 16 hours. At 24 hours postburn, lactated Ringer’s solution is discontinued, and albumin, diluted to 5% in normal saline, is delivered for the subsequent 24 hours.11 Albumin administration is delayed until after 24 hours because of concerns regarding burn edema. This condition may be exacerbated by excessive extravasation of colloid during the first 24 hours when the vasculature is most leaky.
Burns
Burn Injuries
Etiologic Classification of Burns
Thermal Burns
Classification of Thermal Burns
Pathophysiology: the Inflammatory Response
Systemic Response to Thermal Burn Injury
Cardiovascular System: Hypovolemia, Vascular Dysfunction, and Generalized Edema
Cardiovascular System: Myocardial Effects
Gastrointestinal System
Renal System
Hematopoietic System
Immune System
Neurologic System
Metabolic and Endocrine Changes
Burn Treatment
Initial First Aid for Burns
Fluid Resuscitation of the Burn Patient
Fluid Volume
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Burns
Only gold members can continue reading. Log In or Register to continue
