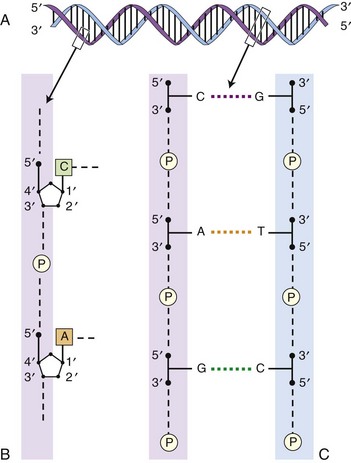
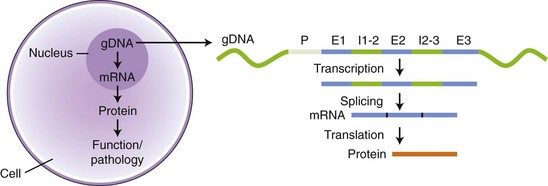
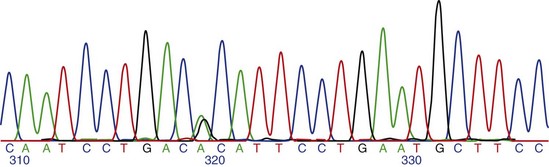
Chapter 2 Thomas Morgan in 1910 identified that genes resided on chromosomes and subsequently demonstrated that they were present at specific locations on chromosomes. The link between genes and proteins was made in 1941 by George Beadle and Edward Tatum, who identified that mutating genes caused changes in specific proteins, indicating that genes and proteins are linked. In 1944, Oswald Avery identified that DNA was the material present in cells that was responsible for heredity, and thus was the material that contained genes. The landmark discovery of the molecular structure of DNA by George Watson and Francis Crick in 195388 solved the conundrum of how genetic information was contained in an organism, and how this information was passed from generation to generation. This discovery enabled development of the field of molecular biology. Deoxyribonucleic acid is composed of four deoxyribonucleotides containing the purine bases adenine and guanine, and the pyrimidine bases cytosine and thymine. In mammalian cells, DNA exists as a double helix, in which two DNA molecules are held together by weak hydrogen bonds to form a DNA duplex (Figure 2-1). Bonding between the two strands of the DNA duplex is restricted by two Watson-Crick rules, specifically, that adenine (A) binds to thymine (T), and that cytosine (C) binds to guanine (G). Therefore, as the two strands of DNA in the DNA duplex are directly complementary, the sequence of one DNA strand can be determined from that of the other. The central dogma of molecular biology was first hypothesized by Crick in 1958 and has subsequently formed the basis of molecular biology teaching. The central dogma states that DNA can be copied to DNA (DNA replication), and that DNA can be copied to messenger RNA (transcription), and that proteins can be synthesized using the information in messenger RNA (mRNA) as a template (translation), but that the information cannot be transferred back from protein to nucleic acid, or from RNA to DNA (Figure 2-2). The structure of RNA differs from that of DNA in a number of ways. The nucleotide base thymine is replaced with uracil, the base pairs are linked by ribose rather than 2′ deoxyribose, and RNA is usually single stranded. RNA is much more susceptible than DNA to degradation by nucleases. Although the genomic DNA sequence does not vary between different cell types, the pattern of message RNA expression is tissue specific. The sequencing of a genome provides the physical map upon which the position of different genes is placed. The human genome sequencing project is widely regarded as one of the great scientific achievements that will have ramifications for humans and other species for years to come. The human genome sequencing project was initiated in 1990 to determine the sequence of base pairs that make up DNA, and to identify the 30,000 genes of the human genome.89 Benefits of sequencing the genome included (1) the expectation that knowledge of the sequence of position of all genes would produce tangible improvements in medical care, (2) that tools could be developed for storing and analyzing the large amount of information produced, and (3) that the work would produce a biotechnology industry to stimulate the development of new medical applications from the data. Such is the importance of the human genome sequence that a parallel, privately funded project was launched in 1998, which aimed to patent the sequence of a selection of genes. However, in 2000 it was ruled that the genome sequence could not be patented and should be made freely available to all researchers. The publicly funded project was completed 2 years ahead of schedule in 2003, and the complete sequence was published. In 2003 the canine genome sequencing project was initiated, funded by the National Institutes of Health. The project was completed in December 2005, and draft sequences covering 99% of the canine eukaryotic genome were published and made publicly available.39 A Boxer Dog was chosen for the canine genome sequencing project because this breed demonstrated the lowest rate of heterozygosity (variation in sequence) when compared with other breeds, thus improving the overall accuracy of the genome sequence and simplifying the genome assembly. The dog genome sequencing project was the fifth large-scale mammalian genome sequence to be published, after those of the human, mouse, rat, and chimpanzee. Just as with the human project, a private company concurrently sequenced a canine genome in parallel with the publicly funded project, using DNA from a male Standard Poodle.35 The canine genome sequence identified nearly 20,000 genes, with most being clear homologues of previously annotated human genes. The canine gene count was less than that reported in the human gene catalogue. Duplication of 216 genes was identified, with most duplicated genes having predicted functions in immunity, reproduction, and chemosensation.39 Expansion of these gene families was interpreted to have resulted from the evolutionary forces of infection and reproductive competition. Extensive analysis of gene sets did not identify any evidence of dog-specific accelerated evolution, although metabolism-related genes were observed to have accelerated more rapidly, suggesting molecular adaptation in carnivores. The publication of an initial feline genome sequence in 2007 covering approximately 65% of the genome of a female Abyssinian Cat has revealed similar insights.55 The feline genome was estimated to be slightly longer (2.7 giga bases) than that of the dog and contains a slightly higher number of genes.56 Of particular interest were the large numbers of endogenous retrovirus-like sequences identified; they account for approximately 4% of the feline genome sequence. Expression of genetic information coded in the DNA sequence is primarily a one-way system, as dictated by Watson’s central dogma, namely, that DNA specifies the synthesis of RNA through the process of transcription. Transcription is mediated by a DNA-directed RNA polymerase and occurs primarily in the nuclei of eukaryotic cells, and to a lesser extent in mitochondria. The length of genes is often many times greater than that of the transcribed mRNA molecule, as the coding sequence is contained within genomic DNA in exons, separated by lengths of noncoding nucleic acid termed introns (see Figure 2-2). Genetic information is contained within exons through its linear sequence of nucleotides, in which groups of three nucleotides (base triplets), termed codons, code for individual amino acids. Thus multiple codons in series across the exons determine the linear sequence of amino acids, which make up the encoded protein. The complete gene sequence, including both introns and exons, is transcribed before posttranscriptional splicing removes the intronic sequence (see Figure 2-2). Translation of mRNA molecules to a polypeptide takes place in the ribosomes. Ribosomes bind to the mRNA molecule at the start codon (AUG) and initiate translation in a 5′ to 3′ direction until a stop codon (UAA, UAG, UGA) is reached. The notation 5′ or 3′ indicates the directionality by naming the carbon atoms in the nucleotide ring (see Figure 2-1, B). Conventionally, nucleic acids can be synthesized in vivo only in a 5′ to 3′ direction, as the polymerase used to assemble new strands can add a new nucleotide only to the 3′-hydroxyl group of the existing nucleic acid sequence. The presence of intronic sequence permits alternative splicing of the exons and thus variation in the sequence, which is translated to protein from a single gene. These splice variants permit different forms of an individual gene from the genomic DNA, which may have differences in function. The functional significance of these changes in relation to disease is yet to be well defined for most conditions, with the exception of tumor biology. An example of the importance of splice variants in the clinical behavior of tumors has been reported with the urokinase-type plasminogen activator receptor (uPAR) gene in breast cancer. Increased expression of a splice variant of the uPAR, lacking exons 4 and 5, is strongly associated with a shorter time to tumor metastasis and a reduction in overall survival.36 This gene has roles in proteolysis and in the induction of cellular proliferation, and a splice variant is hypothesized to confer biologic activity through the loss of a protease-sensitive sequence, which would normally be used for its regulation.36 The clinical importance of the variant is that quantification of the uPAR deletion variant in breast cancer samples can be used as a prognostic measure. The majority of genomic DNA present within mammalian cells is not transcribed, with less than 2% of the haploid human genome coding for genes. The precise function of non–gene coding DNA is unknown, but the hypothesis that this sequence is somehow redundant or unimportant is gradually being disproved. Areas of noncoding elements, which are highly conserved between mammalian species, are often associated with genes that code for regulation of development.39 Marked conservation suggests that these regions are involved in the regulation of gene expression, possibly through their influence on chromatin structure and its relation to the development or maintenance of a cellular state.39 Transcriptionally inactive chromatin has a highly condensed conformation, whereas transcriptionally active chromatin forms a more open conformation. The principle of the one-way flow of genetic information as stated by the central dogma is not without exception. Mammalian genomes contain nonviral DNA sequences, which encode for reverse transcriptase, a protein that can generate a DNA sequence from an RNA template. Reverse transcriptase is utilized by sequences of DNA, termed retrotransposons, which can move around the genome of a single cell. Retrotransposons are transcribed to mRNA in the normal manner, then back to DNA using reverse transcriptase. The DNA can be integrated back into the genome, and this may result in mutations and changes in the quantity of DNA in a cell. Examples of retrotransposons are long terminal repeats, which are similar to retroviruses, short interspersed nuclear elements (SINEs), and long interspersed nuclear elements (LINEs). LINEs are DNA sequences that code for the reverse transcriptase, preferentially making DNA copies of LINE RNA, which can then be integrated into the genome at a new site. SINEs are DNA sequences of reverse-transcribed RNA molecules less than 500 bp in length, originating from tRNA, rRNA, and small nuclear RNA. The precise benefit of SINEs and LINEs is undetermined, but they may have some beneficial significance when incorporated into novel genes to evolve new functionality.65 LINEs and SINEs account for approximately 11% and 18% of the canine genome and 11% and 14% of the feline genome, respectively.55 Insertion of the sequences into functional DNA, such as coding areas, can result in canine diseases. Lamellar ichthyosis is a disorder of epidermal cornification17 that has been reported to develop in Jack Russell Terriers following insertion of a LINE sequence into intron 9 of the transglutaminase 1 gene (TGM1). This insertion results in loss of activity of TGM1 in affected dogs. Centronuclear myopathy, also termed heredity myopathy, is a generalized myopathy affecting Labrador Retrievers that is characterized by muscle weakness and exercise intolerance. The causative mutation has been identified to be a tRNA-derived SINE positioned in exon 2 of the protein tyrosine phosphatase-like, member A (PTPLA) gene.53 The SINE insertion results in loss of the functional exon in the mature mRNA. The best-described reverse transcriptase in mammalian cells is telomerase, which adds a specific DNA sequence repeat to the 3′ end of DNA in the telomere region at the end of eukaryotic chromosomes. Without telomerase, the telomeres are shortened by 50 to 100 bp after each cell division, until they reach a critically short telomere length, at which point the cell enters senescence. The telomere-shortening mechanisms limit cells to a fixed number of divisions and thus are implicated in ageing and oncogenesis. Telomerase replaces the part of the telomere that is lost and thus is naturally expressed in normal cell types with a highly proliferative potential, such as stem cells. More significant, telomerase expression also represents a near universal marker of malignancy,1 as its expression is a mechanism by which tumor cells can avoid telomeric shortening. Consequently, abrogation of telomerase activity is one of the primary candidates for gene therapy of canine tumors, and experimental inhibition of canine telomerase with RNA interference can inhibit tumor growth in vivo.40 Transcription factors are fundamentally important to development, cell signaling, and the cell cycle. Consequently, they are the target of conventional pharmacologic treatment such as anabolic steroid therapy, or estrogen receptor binding proteins such as tamoxifen. Tamoxifen competitively binds to estrogen receptors on tissue targets, producing a nuclear complex that decreases DNA synthesis and inhibits the transcription of estrogen-responsive genes. Consequently, tamoxifen is widely used for the treatment of estrogen receptor positive breast cancer. The manipulation of transcription factors is providing novel avenues of therapeutic intervention. The combination of four transcription factors (OCT4, SOX2, NANOG, and LIN28) is sufficient to reprogram human somatic cells into functional pluripotent stem cells.95 The transcription factor SOX9 is critical to cartilage formation and can be used to restore changes in the extracellular matrix observed in osteoarthritis cartilage, such as the loss of proteoglycans and type II collagen.18 Analysis of transcription factor binding sites in the genome allows the computational modeling of gene regulation. Genes differentially expressed in canine osteoarthritic articular cartilage contain promoter elements that are shared with other higher vertebrates, such as the mouse, rat, and human. This suggests commonality between the transcription factors regulating the changes in gene expression observed in osteoarthritic cartilage. In turn, this implies that the coordinated regulation of chondrocyte differentiation and extracellular matrix reorganization observed in osteoarthritis may be shared between different species.31 Transcriptional control is fundamental to the identity of the cell, as the genomic DNA sequence is identical between different nucleated somatic cells within the same organism. The different phenotypes of cells are conferred by the relative proportion of genes expressed, as this varies dramatically between tissue types and is primarily mediated by regulatory proteins such as transcription factors and signaling molecules.26 Certain genes such as ribosomal proteins and histones have a common function between different cells and thus are constitutively expressed between different cell types; these are termed housekeeping genes. Other genes may demonstrate expression that is largely restricted to particular differentiated cell types, such as type II collagen, which is primarily expressed by chondrocytes. The quantity of gene expression from X chromosomes is regulated, so that for somatic cells it is similar between males, who contain one copy of the X chromosome, and females, who contain two copies. The black and orange alleles of feline fur coloration reside on the X chromosome. Thus in tortoise shell cats, inactivation of the maternal or paternal X chromosome within the skin is evidenced by the hair color.41 Other examples of epigenetic effects include the imprinting of genes, which is the expression of only a single allele of a gene of the two copies inherited from parents, rather than both copies. The copy expressed is determined by which allele is inherited maternally or paternally. Imprinting is estimated to occur in less than 1% of genes.92 Teratogenesis is the interference in normal embryologic development by exogenous factors. An estimated 10% of human birth defects are caused by prenatal exposure to a teratogen. Perhaps the most widely studied is that of thalidomide, which was dispensed as an antiemetic to treat morning sickness between 1957 and 1961. More than 10,000 children are estimated to have been born with birth defects as a result of the teratogenic effects of the drug when given during pregnancy. The molecular basis of the teratogenesis is hypothesized to be the repression of insulin-like growth factor-1 (IGF-1) and fibroblast growth factor-2 (FGF-2) gene expression following thalidomide binding to their promoter sites. Both of these genes stimulate angiogenesis in the normal limb bud76; thus the cumulative effect of their repression is truncation of the developing limb, which is a feature of thalidomide-induced teratogenesis. Some of the mechanisms by which epigenetic processes occur have been defined, such as DNA methylation and histone acetylation. Addition of a methyl group to the cytosine base by DNA methyltransferase converts it to 5-methylcytosine. Genes with marked methylation of the cytosine bases are known to be transcriptionally inactive. Hypermethylation has been identified on the inactive X chromosome when compared with the active copy. Conversely, hypomethylation is associated with transcriptional activity, has been heavily implicated in the neoplastic transformation of cells, and has been identified in canine neoplasia such as lymphoma.54 Alteration of methylation appears to have functional significance in other diseases such as osteoarthritis, in which hypomethylation is associated with protease expression.19 The posttranslational modification of amino acids that make up histone proteins can alter both size and shape of the histone spheres, and thus the relative compaction of the chromatin, which is known to affect the manner in which these genes are expressed. The addition of acetyl groups to histone proteins is associated with gene expression. As histones can be carried into each new copy of DNA in daughter cells because DNA is not completely unwound, this mechanism can produce a non–sequence-based effect of gene expression. The most common mutation is the single nucleotide polymorphism, also termed point mutation (Figure 2-3). A single nucleotide polymorphism within a coding sequence that changes the protein sequence or length is termed a nonsynonymous mutation. A missense mutation results in a change to an amino acid codon, which may alter the protein structure and its biologic activity. Alternatively, the mutation may replace the normal amino acid codon with a stop codon, which is termed a nonsense mutation; this leads to the termination of the protein sequence and the truncation of the protein sequence. A synonymous mutation changes the genetic sequence, not the amino acid, at a codon (as multiple different codons can code the same amino acid), and therefore the protein sequence is not changed. The deletion or addition of a single or multiple base pair sequence will change the frame in which the sequence is read by RNA polymerase and is termed frame shift mutation. Deletions account for approximately 21% of all mutations underlying disease phenotypes, whereas insertions and duplications account for approximately 7%, and missense or nonsense mutations account for 59%.8 The frequency of single nucleotide polymorphisms within the canine genome was estimated to be approximately 1 in every 1500 base pairs within a breed, and 1 in every 900 base pairs when compared between breeds. A dense single nucleotide polymorphism map containing 2.5 million single nucleotide polymorphisms (roughly 1 every 1000 base pairs) has been constructed by comparing the original Boxer genome sequence with the sequence of a Standard Poodle and the partial sequence of nine other dog breeds.35,39 Approximately 70% of single nucleotide polymorphisms identified are polymorphic in other breeds of dog, suggesting they are not breed specific and therefore are likely to be useful studies where traits are mapped.34 Linkage disequilibrium was found to extend up to megabases in length in dogs, which is 40 to 100 times greater than that reported in humans.39,79 Conversely, relatively short levels of linkage disequilibrium are observed when compared between dog breeds. The linkage disequilibrium patterns in dogs reflect the two points in canine evolution where the pool of breeding dogs was reduced: domestication approximately 15,000 years ago, and the subsequent formation of dog breeds in the past few hundred years.34 These events are termed bottlenecks, as they resulted in relative restriction of the active genetic pool for a period of time. Marked linkage disequilibrium has also been reported in purebred cats, although its length is reduced when compared with dogs because of their relatively recent domestication. Gene linkage maps are maps of genetic loci at known genetic intervals across the genome. Microsatellite marker sets exist for the canine genome utilizing more than 500 markers, which provides a resolution (the average distance between loci) of approximately 5 centimorgans across the canine genome.66 The lower the physical distance between loci on a linkage map, the less likely it is that a gene causing a phenotypic trait will be subject to recombination relative to its nearest markers during meiosis, and therefore the more likely it is that marker loci will be transmitted with the trait in the next generation of a pedigree. If the genetic distance between a marker allele and the mutation is small enough that the mutation is transmitted with the trait between generations, they are considered to be in linkage. Linkage can be calculated if all the marker loci on a linkage map are genotyped in each individual in a pedigree, and if each individual is also assessed for the trait (phenotype). A mathematical measurement of linkage with the trait is calculated for each marker locus on the linkage map, and thus the loci in significant linkage with the phenotype can be identified. Although the gene linkage approach is time and labor intensive (requires the genotyping of a large number of loci and the recording of a large amount of phenotypic information), it is the most accurate method for identification of genes involved with a phenotypic trait. The chance of identifying a positive association with such a study is dependent on the quality of the pedigree, the phenotypic information, and the detail of linkage map used. The method works extremely well for monogenetic traits, but polygenic disorders are difficult to elucidate using conventional linkage analysis, as the linkage maps available often are not powerful enough to detect the small effects of the multiple genes involved with the trait. Linkage studies identify linkage to relatively large chromosomal regions, so the identity of the genes responsible for a given disorder requires further study with finer linkage maps and larger pedigree sizes.71 When genes have small effects on a trait, extremely large pedigree numbers are required to produce reliable results.61 The success of microsatellite marker scans for linkage in canine pedigrees has resulted in identification of the genetic basis for a number of monogenetic disorders, such as exercise-induced collapse, which is caused by a mutation in the dynamin 1 gene, a GTPase involved in synaptic vesicle formation.51 Genetic linkage has also been used to evaluate more complex canine traits. A number of traits of canine hip dysplasia such as acetabular osteophytosis,14 hip osteoarthritis,44 hip laxity,82 and radiographic hip score43 have been linked to candidate genomic regions.
Molecular and Cellular Biology
Genomics
Genetics
Genes
Gene Identification
Gene Structure
Control of Gene Expression
Epigenetics
Genomics
Genetic Mutations
Gene Linkage
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


