and Donna L. Senger1
(1)
Hughes Childhood Cancer Research Program, Department of Oncology, Southern Alberta Cancer Research Institute, University of Calgary, Calgary, AB, Canada
Abstract
Glioblastomas are one of the deadliest and most invasive cancers in humans. This heterogenous population of tumours interact with the surrounding brain parenchyma, disrupting physical barriers such as basement membranes, extracellular matrices and cell–cell contact while activating cellular processes that enable infiltration of cells long distances away from the original tumour mass. The protocols describe in this chapter aim to establish experimental models, both in vitro and in vivo, as tools for discovery and evaluation of the underlying molecular mechanisms of cellular invasion by providing experimental methodology that recapitulate, as close as possible, the tumour microenvironment in humans. Although established for the assessment of glioma migration and invasion, these methods provide experimental platforms that can be easily adapted for different types of cancers and for the screening of candidate cancer therapeutics.
Key words
Human gliomaBrain tumoursInvasionMigrationOrthotopic brain tumour modelsMurine model of human glioma1 Introduction
1.1 Disease Reservoirs of Malignant Glioma
Human malignant glioma (MG) is one of the most common primary central nervous system tumours in adults with a median survival of 12–14 months and long-term survivors (i.e. surviving ≥ 3 years) are rare (1–3). A major barrier to effective malignant glioma treatment is the highly invasive nature of these tumours, where tendrils of tumour extend into the normal surrounding brain parenchyma rendering these tumours incurable by local therapies such as surgery or radiotherapy (4–6). Specifically, the tumour cells activate a number of coordinate cellular programs including those necessary for migration and invasion (7). These cells, along with a second highly invasive cell population, the recently described “brain tumour initiating cells” (BTICs), act as “disease reservoirs” which elude our current treatments and ultimately lead to disease relapse (8–14). Although the exact characterization of the BTIC cell population is still debated (15), these cells, when implanted into the brains of immunocompromised mice, result in a highly invasive and tumourigenic tumours that closely resemble the human disease (10, 16, 17). The invasive traits of these two cell populations together with the ability of invading glioma cells to reduce proliferation and upregulate survival pathways (5, 18–20) makes them highly resistant to conventional treatments (11, 12, 20). Identifying the biological properties of these distinct populations of cells within malignant glioma and specifically the underlying invasive mechanisms will aid in the development of new effective treatments (21).
1.2 Molecular Aspects of Glioma Progression and Invasion
Glioma invasion is not random but rather occurs along white matter tracts (optic radiations, corpus callosum or anterior commissure), basement membranes of blood vessels, along the subependyma and adjacent to neurons. In contrast to other organs, there are few “classical” extracellular matrix (ECM) proteins (collagen, laminin, vitronectin, fibronectin) in the brain except around blood vessels and the glia limitans externa. Instead the ECM of the brain is mostly composed of glycosoaminoglycans (GAGs) and a few structural glycoproteins, with the main GAGs being hyaluronic acid (HA), chondroitin, keratin and heparan sulphates (22). In addition, the glioma cells themselves synthesize many ECM components including tenascin, vitronectin, collagen types I, III, IV and VI, fibronectin, laminin, hyaluronan, chondroitin sulphate and heparan sulphate proteoglycans.
Over the years many individual genes have been implicated in glioma invasion and recently gene expression profiling has identified a subclass of gliomas expressing several genes with specific roles in cell migration and invasion that strongly correlate with poor patient survival (23–26). At present, no comprehensive model of glioma invasion exists; however, invasion can be conceptualized into discrete steps that include adhesion to the ECM, degradation of the ECM to allow passage, and glioma cell movement. Several cell surface receptors have been implicated in glioma adhesion and invasion, including the integrins (27–30), immunoglobulin superfamily members (e.g. NCAM) (4), cadherins (4, 31), CD44 (32, 33) , BEHAB (34, 35), elastin binding protein (36), SPARC (37), the ephrin (Eph) receptor–ligand system (38–41), semaphorins (42, 43), plexins (43) and the neurotrophin receptor p75NTR (44, 45). In addition to activating downstream signal cascades important for cell movement, engagement of many of these receptors results in the activation of several classes of proteases including matrix metalloproteinases (MMPs), ADAMs (a disintegrin and metalloproteinase), plasmin and cathepsins reviewed in (46–48) necessary for the degradation of the ECM.
1.3 Development of Assays to Evaluate Glioma Invasion
As a result of intense investigation in the area of cell invasion, numerous techniques have been establishment to identify molecular events required for the invasive ability. In the area of glioma invasion, strategies to directly identify genes and proteins important for invasion have included isolation of invasive cells from human cell lines in vitro (18, 19), the use of organotypic brain slice cultures (36, 49–53) and the collection of tumour and invasive cells from the main tumour mass in frozen glioblastoma patient specimens using laser capture microdissection (54–57). In addition, in vitro assays have been generated to assess the role of individual proteins or processes in glioma invasion, many of which will be discussed in this chapter. Although these techniques have been used successfully, no single technique is sufficiently comprehensive and the selection of assay(s) performed in a given study should be chosen with care. Here we will outline several systems that we have previously used to either verify or identify the underlying mechanisms involved in glioma invasion both in vitro and in vivo.
2 Materials and Methods
2.1 In Vivo Selection of Highly Invasive Glioma Variants
Background
One of the problems in xenotransplanting human glioma cells into the brains of immunocompromised mice is that the resulting tumours are circumscribed with very little cell infiltration into the brain parenchyma (58) (see Fig. 1, for example). To generate an orthotopic model that more closely mimics the human disease and allows for the identification of molecular determinants of glioma invasion in a global and unbiased manner, we hypothesized that using an in vivo selection strategy similar to the one developed by Richard Hynes and colleagues for metastatic melanoma (59), we could develop an in vivo glioma model that more closely recapitulates the human condition thus providing a strategy to isolate genetically and phenotypically distinct population of glioma cells that are highly invasive. As proof of concept, using the in vivo selection strategy outlined below, we previously isolated highly invasive glioma variants from two established human glioma cell lines U87MG and U251N (44). Stable transfectants of U87MG and U251N expressing green fluorescent proteins (GFP) and a neomycin resistant gene were serially transplanted into the brain of immunocompromised mice. Cells from the main tumour mass, and those that migrated to the contralateral hemisphere were isolated, grown and re-injected into the brains of a second mouse. Using this approach, both highly tumourigenic and highly invasive populations of cells were isolated and characterized for their phenotypic and genotypic changes (44).
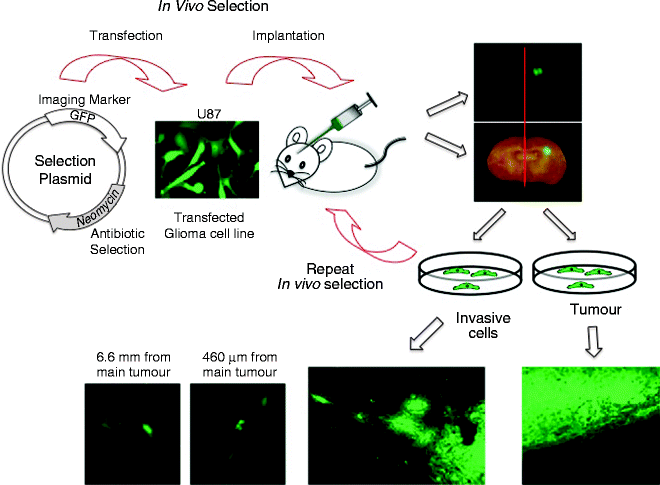
Fig. 1.
Schematic of an in vivo selection strategy for the isolation of highly invasive glioma variants from an established glioma cell line. (Top panel) Transfect a non-invasive human glioma cell line with a plasmid containing an imaging marker (e.g. GFP) to visual the tumour cells in vivo, and a drug resistance or selection gene (e.g. Neomycin) to selectively isolate and grow the tumour cells in culture. Implant cells into the putamen of the brain of an immunocompromised animal using a stereotactic apparatus. Allow the tumours to establish for 4–6 weeks at which time the animals are sacrificed and the ipsilateral side of the brain containing a grossly visible tumour is separated from the contralateral side containing only isolated invasive glioma cells. Dissociate the brain tissue to single cells. Grow the cell suspension in media containing the appropriate antibiotic for the plasmid drug selection, then re-implant the isolated tumour cells into a second immunocompromised animal repeating the process to select for increasingly non-invasive or invasive glioma cells. The ipsilateral side of the brain contains a grossly visible tumour with a well demarcated border (right lower panel) while the tumour cells isolated from the contralateral side form tumours with highly infiltrative borders, with individual cells migrating substantial distances from the main tumour mass (lower left 3 panels).
Materials
All cell culture protocols outlined in this chapter use DMEM Complete Media that contains Dulbecco’s modified Eagle’s medium F12 supplemented with 10% heat-inactivated foetal bovine serum, 1% antibiotic/antimycotic, 0.1 mM non-essential amino acids, 2 mM l-glutamine and 1 mM sodium pyruvate (Gibco BRL, Gaithersburg, MD, USA). The choice of culture media may vary depending on the cell line being assessed; thus, the DMEM Complete Media should be substituted with the appropriate media for each individual cell line used. All other products used are included in the methods outlined below. Comparable products from other suppliers should also be effective.
2.1.1 Establishment of Invasive Variants from a Human Glioma Cell Line by In Vivo Selection
1.
Select a human glioma cell line that has the ability to establish intracranial tumours in immunocompromised mice (e.g. U87MG or U251 obtained from ATCC). Grow and maintain cells using standard tissue culture protocols in DMEM Complete Media or the appropriate media for each individual cell line depending on the growth requirements, at 37°C in a humidified 5% CO2 incubator.
2.
Establish stable cell lines expressing a selection plasmid that allows for the re-isolation of the glioma cells once they have been passaged through the animal. Choose a selection plasmid that expresses both GFP (to visualize the implanted glioma cells) and an antibiotic resistance gene (e.g. neomycin for recovery of the tumour cells post-intracranial implantation via antibiotic selection) such as the pEGFP-N1 vector from Clontech (Palo Alto, CA, USA). Seed cells at 2 × 105 cells/well in a 6-well tissue culture plate in DMEM Complete Media and incubate overnight at 37°C in a humidified 5% CO2 incubator. Introduce the pEGFP-N1 vector using FuGENE™ 6 transfection reagent (Roche Diagnostic Co., Indianapolis, IN, USA) according to the manufacturers instructions. The plasmid and FuGENETM reagent can be combined in various ratios in serum-free media and incubated at room temperature for 30 min to allow for the formation of transfection complexes. Add the plasmid/FuGENETM mixture drop wise to the cells and incubate the cells in a humidified 5% CO2 incubator at 37°C overnight.
Note: Using a linearized plasmid usually helps increase the efficiency of establishing a stable cell line.
3.
The following day change the media to fresh DMEM Complete Media containing G418, or the appropriate drug selection antibiotic for the transfection plasmid used, to select for cells that have taken up the vector. Determination of the appropriate concentration for the selection drug should be determined prior to expression vector transfection by performing a drug sensitivity curve for each cell line, selecting the minimum concentration of drug that gives 100% cell death (see below, Sect. 2.1.2); usually in the range of 100–400 μg for established glioma lines. Grow the cells under antibiotic selection until the wells are confluent. GFP-transfected cells can be identified by fluorescent microscopy and GFP expression of >95% can be obtained by fluorescence-activated cell sorting (FACScan flow cytometer; Becton, Dickinson and Company, Oakville, ON, Canada).
4.
Stable transfectants of glioma cells can be maintained in the same media, with the addition of G418 at the concentration determined above (Invitrogen, Carlsbad, CA, USA). It is important to have stable clones prior to implantation in the animals so that the in vivo selected glioma cells maintain expression of the plasmid.
5.
For in vivo implantation, harvest actively growing GFP/neomycin glioma cells using trypsin–EDTA (0.25%), wash twice in sterile PBS [137 mM NaCl, 8.1 mM Na2HPO4, 2.68 mM KCl and 1.47 mM KH2PO4 (pH 7.5)] and resuspend 107 cells in 100 μl PBS.
6.
For the stereotactic injection, use a 10-ml syringe (801RN Hamilton Co, Lot#436284) with a 30-gauge needle (RN NDL Lot#440603) mounted on a Kopf stereotactic apparatus (Model 900 Small Animal Stereotaxic Instrument, Kopf Instruments, Tujanga, CA; http://www.kopfinstruments.com) outfitted with a mouse adaptor (Model 926 Mouse Adaptor includes a nose and tooth (palate) bar assembly with zygoma ear cups).
7.
Anesthetize the mouse using an intraperitoneal administration of ketamine 200 mg/kg and xylazine 10 mg/kg (MTC pharmaceuticals, Cambridge ON, Canada) in sterile water. In addition, give a subcutaneous pre-operative dose of buprenorphine 0.05 mg/kg for pain. Implant 1 × 105 cells/mouse in 2–3 μl sterile PBS, intracerebrally into the right putamen of 6- to 8-week-old female SCID mice at a depth of 3 mm through a scalp incision and a 0.5 mm burr hole made 1.5–2 mm right of the midline and 0.5–1 mm posterior to the coronal suture. To ensure the cells are injected into the putamen, the head position should be flat or parallel to horizontal, if available, use ear bar stabilizers (Kopf Instruments, Model 921 Zygoma Ear Cups) to maintain correct head position. The implantation depth of 3 mm is the depth after the needle has entered the brain tissue. Once a depth of 3 mm has been reached, go a further 1 mm forward, then withdraw the needle back 1 mm to the original 3 mm depth; this provides additional space for the cells to be deposited. To prevent the withdrawal of tumour cells along the needle tract, inject cells slowly (30 s to 1 min), wait 3–5 min to allow the cells to adhere to the brain tissue and then slowly withdraw the needle allowing the tissue to close behind the needle. It is important to observe any deviation of the implantation site caused by the needle that is not perpendicular to the horizontal, this could result in an implantation site too close to the lateral ventricle.
8.
Once the needle has been withdrawn, the incision is closed using a 9-mm Clay Adams wound clip (BD #427631).
9.
House the animals according to your institutions animal regulations. In our case, animals are housed in groups of three to five and maintained on a 12-h light/dark schedule with a temperature of 22 ± 1°C and a relative humidity of 50 ± 5%. Food and water should be available ad libitum.
10.
Following surgery, weigh and record the body weight of each animal daily and observe the animal for any signs of discomfort.
11.
Allow at least 21 days for the formation of the tumour. The actual duration will depend on the health of the mouse and the type of cells injected.
12.
To isolate the tumour and invasive glioma cell populations, sacrifice the mouse, divide the brain coronally close to the needle tract and examine using fluorescence to visualize the GFP-expressing glioma cells (Fig. 1). Ensure that the main tumour mass is completely embedded within the normal tissue of the brain. This is to make certain that the tumour cells present in the contralateral hemisphere arrived there by invading through the normal brain parenchyma.
13.
For tissue culture, separate the hemisphere containing the main tumour mass from the contralateral hemisphere. Independently isolate cells from each hemisphere as described below (Sect. 2.1.3).
14.
Resuspend the tissue homogenate and gently force through a 100-μm mesh cell strainer (BD Bioscience; BD FalconTM Cell Strainer, Catalogue # 352360) into a 50-ml conical centrifuge tube. The resulting cell suspension is then centrifuged at low speed (400–500 × g) for 10 min.
15.
Remove the supernatant and resuspend the cell pellet in DMEM Complete Media containing G418 (in our case 400 μg/ml) to select for the GFP-transfected tumour cells. Cells obtained from the hemisphere containing the main tumour mass are the “tumour” cells and cells isolated from the contralateral hemisphere are the “invasive” cells. Maintain the culture at 37°C with 5% CO2. Once the cells have reached 80% confluence, passage the cells to expand them prior to implantation into the brain of a second immunocompromised mouse. Depending on the number of invasive cells recovered from the contralateral hemisphere, the invasive cultures may need to be maintained and expanded for 2–3 months to obtain enough cells to be re-inoculated into a SCID mouse. Repeat the procedure until two distinct in vivo tumour populations are observed.
After 2–3 rounds of in vivo selection, the brains of the immunocompromised mice implanted with the non-invasive “tumour” cells should form large tumours with sharply demarcated edges (Fig. 1; lower right) while the brains of mice implanted with the “invasive” cells should form highly infiltrative tumours with poorly defined edges (Fig. 1; lower left). Using this experimental strategy, we have previously isolated two populations of cells from a single parental cell line that are phenotypically and genotypically distinct with gene expression differences between the non-invasive and highly invasive in vivo selected glioma cells (44).
2.1.2 Determination of G418 Sensitivity
1.
Plate 2 × 104 cells/well into a 6-well plate (10 wells total).
2.
Grow cells for 24 h at 37°C in a humidified 5% CO2 incubator.
3.
Remove media and then add media containing varying concentrations of G418 (0, 50, 100, 200, 300, 400, 500, 600, 800, 1,000 μg/ml).
4.
Replenish the selection medium every 3–4 days and observe the percentage of surviving cells over time.
5.
Select the minimum concentration that kills 100% of the cells in 2 weeks (For U87MG and U251N, we have determined that 400 μg/ml of G418 is optimal).
Similar strategy can also be used for other selection drugs.
2.1.3 Preparation of Tumour and Invasive Cells from Mouse Brain Injected with GFP/Neomycin Transfected Human Glioma Cells
The following experimental procedure should be performed under sterile tissue culture conditions.
1.
Once the brain has been removed, wash the brain tissue three times in sterile PBS in 60-mm petri dish by transferring from one dish to another using a sterile 18-gauge needle.
2.
Transfer the tissue into a clean 60-mm dish containing 2 ml of sterile PBS.
3.
Holding the tissue with a needle, use a sterile scalpel to mince the tissue into small pieces.
4.
Transfer all of the tissue into a 15-ml conical tube using a 10-ml pipette.
5.
Add 500 μl of 10× trypsin–EDTA (2.5% stock; Invitrogen; Catalogue # 15400-054) and 500 μl of 1 mg/ml DNAse I (Sigma; Catalogue # D5025) to the tissue suspension.
6.
Incubate the tissue suspension in 37°C water bath for 30 min, mixing occasionally.
7.
Place 100-μm mesh cell strainer (BD Bioscience; BD FalconTM Cell Strainer, Catalogue # 352360) on top of a 50-ml conical centrifuge tube. Resuspend the tissue homogenate and gently force the homogenate through the mesh using the plunger of a 3-cc syringe into the centrifuge tube.
8.
Wash the mesh strainer four times with 4 ml of DMEM Complete Media, continuing to gently force the tissue through the mesh with the syringe plunger.
9.
Centrifuge the cell suspension at low speed (400–500 × g) for 10 min to pellet cells.
10.
Remove the supernatant and resuspend cells in 12 ml of DMEM Complete Media containing G418, or the appropriate selection drug for the plasmid used, at the determined concentration (e.g. 400 μg/ml for U87MG).
11.
Plate tumour cells in a 100-mm tissue culture dish in DMEM Complete Media containing G418.
12.
Place the cultures in a tissue culture incubator at 37°C for 2 days.
13.
Replace 2/3 of the volume of media with fresh media containing G418 (e.g. 400 μg/ml for U87MG) and return to the tissue culture incubator.
14.
Replace all media with fresh media containing G418 after 1 or 2 days.
15.
Replace media every 2–3 days to keep G418 active.
16.
Confirm the presence of GFP expressing cells under fluorescent light.
17.
Expand the cells until there are enough cells to inject a second set of immunocompromised mice [recommend injecting 4–5 mice (1 × 105 cells/mouse) for each round of in vivo selection].
2.2 Assessing Glioma Migration In Vitro
2.2.1 In Vitro Circular Migration Assay
Migration assays described here are performed using a microliter-scale radial monolayer assay adapted from Berens et al. (60).
Equipment
10-Well Teflon-masked microscope slides (Creative Scientific Methods Phoenix, ZA, USA); autoclaved
Cell Sedimentation Manifold (Creative Scientific Methods, http://www.creative-sci.com, Phoenix, AZ, USA)
Hemocytometer
Image analysis software
2.3 Method
1.
Coat 10-well Teflon-masked microscope slides with 50 μl of 20 μg/ml laminin (Sigma-Aldrich) or ECM of choice, for 1 h at 37°C. ECM chosen will dependent on the cell line used.
2.
Remove laminin and wash the microscope slide once with 100 μl of sterile phosphate buffered saline.
3.
Remove cells from the tissue culture dish by rinsing once with sterile PBS followed by Puck’s EDTA (1 mM EDTA, 10 mM HEPES, 5 mM KCl, 140 mM NaCl, 4 mM NaHCO3, and 6 mM Dextrose, pH 7.3).
Note: Cells are released from the culture dish using Puck’s EDTA to prevent cleavage and activation of cell surface receptors (e.g. the family of proteinase-activated receptors) that may occur with Trypsin–EDTA.
4.
Incubate cells at 37°C for ~5 min.
5.
Collect cells by washing the tissue culture dish containing the cells with DMEM Complete Media and placing the cell suspension in a 15-ml conical centrifuge tube.
6.
Centrifuge cells at 400–500 × g for 5 min to pellet the cells, then remove the supernatant. Resuspend the cell pellet in a minimal amount of DMEM Complete Media and count cells using a hemocytometer.
7.
Prepare a cell suspension of 1,000 cells/μl media (2 μl of cell suspension is required per well).
8.
Place the Cell Sedimentation Manifold (CSM Inc., Phoenix, AZ, USA) over the laminin-coated slides to establish a circular 1-mm diameter confluent monolayer (Fig. 2).

Fig. 2.
Shown is the Model 900 Small Animal Stereotaxic Instrument from Kopf Instruments (http://www.kopfinstruments.com) outfitted with a mouse adaptor [Model 926 Mouse Adaptor with a nose and tooth (palate) bar assembly with zygoma ear cups]. The stereotactic apparatus is outfitted with a 10-ml Hamilton syringe (blue; 801RN Hamilton Co) with a 30-gauge needle.
9.
Add 50 μl of media to each well of the sedimentation manifold.
10.
Seed 2 μl (2,000 cells) into the central lumen of the sedimentation cylinder (5 wells per cell type/culture condition or treatment).
11.
Place slides on ice for 60 min and then place the sedimentation manifolds at 37°C in a humidified 5% CO2 incubator for ~6 h.
12.
Confirm that the cells have completely attached to the slide and remove the sedimentation manifold.
13.
Remove the media and incubate in fresh media plus/minus any additional experimental treatments.
14.
Using a phase contrast or fluorescent (if labelled with a fluorescent marker) microscope, take a digital image of each well as the starting point of the assay (before migration).
15.
Acquire digital images at desired time points.
16.
Measure the radius of the best-fit circles using image analysis software (e.g. Axiovision 4.2 imaging software; Carl Zeiss Canada Ltd.) around the area covered by cells (illustrated in Fig. 2).
17.
Calculate the quantitative migration scores as the increase of the radius beyond the initial radius of the object.
2.4 Monitoring Glioma Invasion In Vitro
2.4.1 In Vitro Invasion Assay
Equipment
The assays described in this section use a traditional transwell insert for a 24-well plate (6.5-mm diameter) with an 8.0-μm pore; however, several membrane diameters and pore sizes are available, but for mammalian cells that are not neutrophils, a pore size of 8.0 μm is needed to allow the nucleus to move through the membrane. The self-centering hanging design keeps the membrane about a millimetre off the bottom of the well.
Method
1.
The day prior to setting up the in vitro invasion assay, remove culture media from cells to be used, wash with sterile PBS and replace with serum-free media. Incubate cultures overnight at 37°C in a humidified 5% CO2 incubator.
2.
Prepare MatrigelTM to be used by diluting one part MatrigelTM (BD Biosciences, Catalogue # 354234) with two parts cold serum-free media using cold pipette tips (−20°C).
3.




Insert an 8-μm pore size transwell invasion chamber (BD Bioscience, Mississauga, ON, Canada) (Fig. 3) into a 24-well tissue culture dish containing 500 μL of matrix to wet the outside of the membrane, removing any excess liquid. Layer 30 μl of MatrigelTM into the cup of the transwell and let dry in a tissue culture hood. To avoid the formation of a meniscus and to get a more even layer of MatrigelTM, a layer of sterile ddH2O can be placed on top of the MatrigelTM before letting it dry. The transwells can be stored 4°C for up to 1 week. Alternatively, transwells can be coated with ECM of choice; e.g. 20 μg/ml laminin, PurecolTM collagen mix (#5005-B) used at nine parts collagen to one part 10× DMEM Complete Media, or brain-tumour-like-matrix (see Sect. 2.4.4).
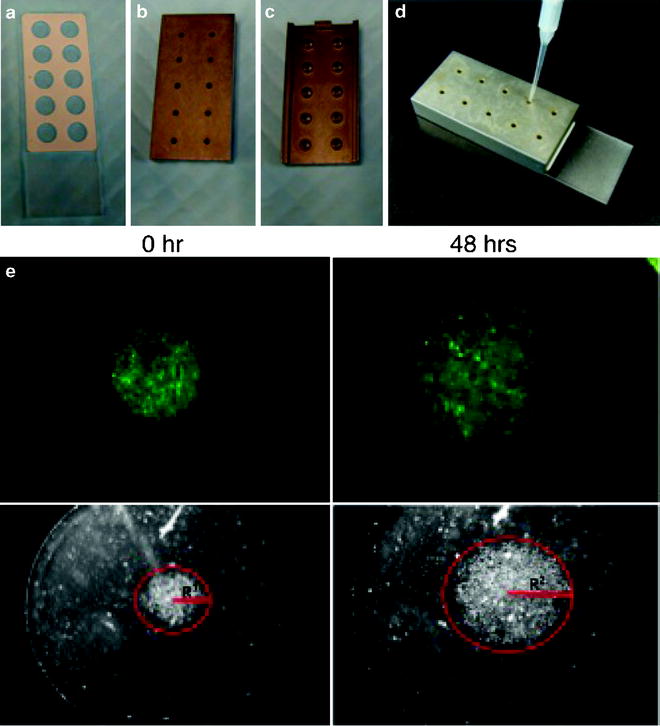
Fig. 3.
Images in (a)–(d) show the components of the circular migration assay that contains a 10-well Teflon-masked microscope slide (a), and the Cell Sedimentation Manifold, upper, lower and side view (b–d). Images shown in (e) are fluorescent micrographs (top panel) and photomicrographs (lower panel) of U87MG glioma cells expressing GFP plated in 10% FBS and grown on 20 mg/ml Laminin. Images on the left were taken immediately after the removal of the Cell Sedimentation Manifold while images on the right were taken 48 h later. Distance travelled by the glioma cells is measured as the difference in the radius between the two time points (R2 − R1; red). The time of the assay can be modified based on the migratory rates of the cells to be assessed.
Note: As different cell lines and different treatments can result in a wide range of invasive potential, it is important when choosing a matrix to be aware of the matrices properties. For example, MatrigelTM is a basement membrane (contains collagen IV and laminin) and will cause polarization/differentiation of the test cells. Cells in this state will have decreased migration providing a more rigorous measure of invasion. Collagen I/III can be used as a more general representation of stromal ECM allowing one to measure the movement of cells without having to overcome the integrin polarization signals, or the more specific “brain-tumour-like-matrix” described for the 3D invasion assay (Sect. 2.4.4) can be used to more closely mimic the in vivo tumour environment.
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
