Chapter 61 Liver
Structure and Function
Liver Structure
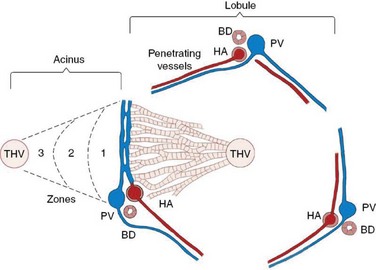
The hepatic lobule is the anatomic unit of the liver. In the anatomic model, liver lobules are organized into irregular polygons demarcated by connective tissue and composed of plates of hepatocytes radiating outward from the central vein to the portal triads (Figure 61-1). The hepatic acinus is the functional unit of the liver. In the functional model hepatocytes are instead oriented around the afferent vascular system (portal veins and hepatic arteries) just as they anastomose into sinusoids (Figure 61-1), and the central vein is at the periphery of the acinus instead of centrally located as in the anatomic model. The acinus is divided into three contiguous zones (1, 2, and 3) that correspond to distance from the arterial blood supply. Those hepatocytes in closest proximity to the arterioles (zone 1) receive the greatest oxygen content, but are also first in line to be affected by toxins transported from the gut to the portal vein. Zone 3 hepatocytes reside at the periphery of the acinus near the central vein, and zone 2 hepatocytes are interspersed between zone 1 and zone 3 hepatocytes. The anatomic model is perhaps easier to understand, but the functional model serves as a better foundation for understanding liver pathology.1 In either model portal venous and arterial blood flow centripetally, that is, toward the central vein, whereas bile flows centrifugally, that is, away from the central vein. Hepatocytes extract nutrients and oxygen from portal and arterial perfusion, respectively, and produce bile acids and other bile constituents that are transported from hepatocytes into bile canaliculi, ductules, and ducts.
Biliary Tract Structure
The basic elements of the biliary tract are the hepatic canaliculi, bile ductules, intralobular ducts, interlobular ducts, hepatic ducts, cystic duct, gallbladder, common bile duct, and the pancreaticobiliary sphincter (of Oddi).2 There are many variations on this central theme, the most important of which are (a) the pancreaticobiliary sphincter is a common physiologic and anatomical channel at the duodenal papilla in the cat3 and (b) there are many anatomic variations in the feline gallbladder, from single gallbladder to bilateral gallbladders, body duplication, fundic duplication, complete duplication, septate, and Y-shaped gallbladder.4
Cells of the Liver
Hepatocytes
Hepatocytes account for 60% to 80% of the liver cell mass (see Table 61-1) and contribute to a wide range of metabolic activity, including carbohydrate, protein, lipid, nucleic acid, porphyrin, metal, vitamin, glutathione, hormone, and xenobiotic metabolism; coagulation factor synthesis; biliary secretion; and immune surveillance.1,5 Hepatocytes have an eosinophilic cytoplasm reflecting numerous mitochondria, and basophilic stippling caused by large amounts of rough endoplasmic reticulum and free ribosomes. Hepatocyte nuclei are round with dispersed chromatin and prominent nucleoli. Anisokaryosis is common and often reflects various degrees of polyploidy, a normal feature of more than 50% of hepatocytes. The average life span of the hepatocyte is 5 to 6 months reflecting their ability to regenerate. Hepatocytes are organized into plates separated by vascular channels (sinusoids), an arrangement supported by a reticulin (collagen type III) network. The sinusoids have a discontinuous, fenestrated endothelial cell lining. The endothelial cells have no basement membrane and are separated from the hepatocytes by the space of Disse, which drains lymph into the portal lymphatics. Hepatocytes are supported by a number of other cell types, which account for 40% of the liver cell mass.
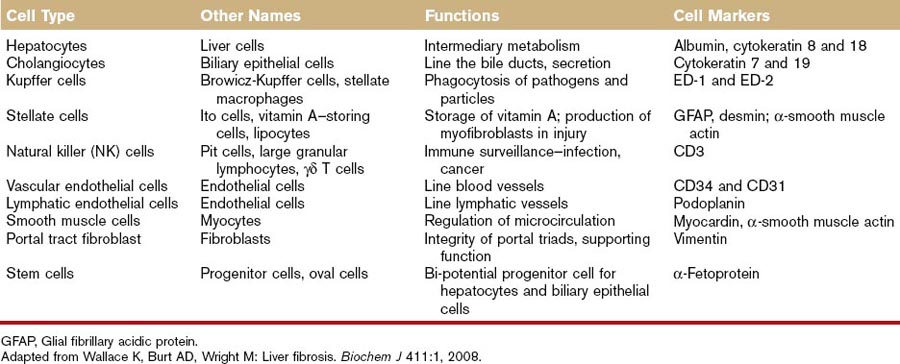
Cholangiocytes
Representing 3% to 10% of liver cell mass, cholangiocytes are also known as biliary epithelial cells.6 They secrete water, bicarbonate, and cations into the bile in the physiologic state, but they may also participate in the immune response as antigen-presenting cells in disease states. The biliary tract is a convergent system of canals that begins in the canaliculi, followed by the bile ducts, and ending with the common bile duct. Bile secretion depends on the function of membrane transport systems in hepatocytes and cholangiocytes and on the structural and functional integrity of the biliary tract. The hepatocytes, constituting the most abundant liver cell population, generate the so-called primary bile in their canaliculi. Biliary canaliculi are blind tubular structures, with a very high surface-to-volume ratio that by means of osmotic gradients favors the formation of bile flow. Cholangiocytes, which constitute 3% to 10% of the liver cells, modify the canalicular bile by secretory and reabsorptive processes as bile passes through the bile ducts, and they are responsible for approximately 30% of bile volume. In contrast to hepatocytes, where secretion is constant and poorly controlled, cholangiocyte secretion is broadly regulated.5,6
Kupffer Cells
In disease states, Kupffer cells contribute to the pathology of ethanol and other toxic principles through production of inflammatory mediators, activation of Toll-like receptors, and elaboration of tumor necrosis factor (TNF-α).7 Kupffer cell activation is responsible for early ethanol-induced liver injury, common in chronic alcoholics. Ethanol activates the Toll-like receptor 4 and CD14, receptors on the Kupffer cell that internalize the endotoxin lipopolysaccharide. Internalization activates the transcription of TNF-α and production of superoxide (a prooxidant). TNF-α then enters the stellate cell in the liver, leading to collagen synthesis and fibrosis. Fibrosis eventually causes cirrhosis or loss of function of the liver (see the role of the stellate cell, which is discussed in “Stellate Cells” section that follows).
Stellate Cells
Hepatic stellate cells (HSCs) (also referred to as vitamin A–storing cells, lipocytes, interstitial cells, fat-storing cells, and Ito cells) exist in the space between parenchymal cells and liver sinusoidal endothelial cells of the hepatic lobule and store 50% to 80% of vitamin A in the whole body as retinyl palmitate in lipid droplets in the cytoplasm.7–10 In physiologic conditions, these cells play pivotal roles in the regulation of vitamin A homeostasis. In pathologic conditions, such as hepatic fibrosis or liver cirrhosis, HSCs lose vitamin A and synthesize a large amount of ECM components, including collagen, proteoglycan, glycosaminoglycan, and adhesive glycoproteins. The morphology of these cells also changes from that of the star-shaped stellate cell to that of the fibroblast or myofibroblast. HSCs are now considered to be targets of therapy of hepatic fibrosis or liver cirrhosis.11 Activation of HSCs, a key event in liver fibrosis, is caused by diminished adipogenic transcription.12 Wnt signaling inhibits antiadipogenic activation of HSCs and liver fibrogenesis; wnt antagonism inhibits HSC activation and liver fibrosis.9
Endothelial Cells
Lymphocyte recruitment from the circulation into tissue is dependent on the ability of the lymphocyte to recognize and bind molecules on the endothelial cell surface that promote transendothelial migration. A multistep model of leukocyte adhesion to vascular endothelium has been described and is broadly applicable, although the details of the signals involved differ between tissues.13,14 In a generally accepted model, tethering or rolling receptors expressed on endothelial cells capture free-flowing leukocytes. These receptors may be either selectins or members of the immunoglobulin superfamily. Once captured, the leukocyte can receive activating messages presented by endothelial cells in the form of chemokines that activate specific G-protein–coupled receptors on the leukocyte surface. Occupancy of these receptors triggers a cascade of intracellular signals that results in the presentation of high-affinity integrin receptors on the leukocyte surface that bind to immunoglobulin family of counterreceptors on the endothelium to promote leukocyte arrest on the vessel wall. In the presence of the appropriate migratory signals the leukocyte will migrate across the endothelium into tissue, where it follows a hierarchy of chemotactic signals toward the focus of inflammation.
Stem Cells
It is difficult to arrive at a universally applicable definition of a stem cell because some of the defined properties of a stem cell can be exhibited by the stem cells in some tissues or organisms but not in others. In spite of that, a generally acceptable consensus defines a stem cell as an undifferentiated cell that has capacity to self-renew, for production of progeny in at least two lineages, for long-term tissue repopulation after transplantation, and for serial transplantability. In addition, stem cells exist in a mitotically quiescent form and clonally regenerate all of the different cell types that constitute the tissue in which they exist. They can undergo asymmetrical cell division, with production of one differentiated (progenitor) daughter and another daughter that is still a stem cell. The offspring of stem cells are referred to as progenitor cells, also named as transit amplifying cells and therefore cannot be serially transplanted, and are classified as early and late. The early progenitor or stem/progenitor cells have multilineage potential and similar characteristics to stem cells. The late progenitor cells have differentiated further and produce progeny in only a single lineage. Although they divide rapidly, they are capable of only a short-term tissue reconstitution and they do not self-renew.15
Early studies in hepatocyte turnover and liver regeneration showed that the parenchymal cell, the hepatocyte, was the primary and only cell involved in tissue renewal. However, new studies of liver regeneration, hepatocarcinogenesis, liver transplantation, and various cell lines show that a variety of cell types participate in maintaining hepatocyte number and mass. Recent studies indicate the presence of both intrahepatic and extrahepatic stem/progenitor cell populations that serve to maintain the normal organ and to regenerate damaged parenchyma in response to a variety of insults. The intrahepatic compartment most likely derives primarily from the biliary tract, particularly the most proximal branches, that is, the canals of Hering and smallest ductules. The extrahepatic compartment is at least in part derived from diverse populations of cells from the bone marrow. Embryonic stem cells are considered as a part of the extrahepatic compartment.16 The precise role(s) of each of these individual cells remains to be determined, but it is clear that in the aggregate they confer the vast regenerative capacity of the liver .
Liver Function
Metabolism
The liver is involved in many aspects of intermediary metabolism.1
Carbohydrates
• Early fasting: glycogen → glycogenolysis → glucose → normoglycemia
• Prolonged fasting: amino acids → gluconeogenesis → glucose → normoglycemia
The enzyme responsible for the dephosphorylation of glucose-6-phosphate is glucose-6-phosphatase-α. Alterations in quantity, location, or activity of glucose-6-phosphatase, such as those seen in type 1 glycogen storage diseases, effectively result in a lack of all endogenous glucose production and severe hypoglycemia develops during periods of fasting.17
Proteins
The liver is an important site of protein metabolism. Amino acids and proteins absorbed from the intestine or produced in the body are delivered to the liver. The liver deaminates amino acids and converts them to carbohydrates and lipids.18–21 Deamination produces α-keto acids, which can be metabolized for energy or used for synthesis of monosaccharides and fatty acids.20 The liver synthesizes amino acids from intermediates of carbohydrate and lipid metabolism by amination and transamination.21 Examples of amino acid transaminations include:
Lipids
Exogenous Transport
Triglyceride is the major dietary lipid, along with cholesterol, phospholipids, and fat-soluble vitamins.22 The digestion of dietary lipids begins in the proximal GI tract with the action of lingual and gastric lipases, and is completed in the small intestine with the actions of pancreatic lipase, cholesterol ester hydrolase, and phospholipase A2. Lipid digestion and absorption is more complicated than carbohydrate and protein digestion and absorption because of lipid solubility characteristics, and involves emulsification of lipids by bile salts, hydrolysis by pancreatic lipase and colipase, solubilization of fatty acids and monoglycerides into mixed micelles, absorption, reesterification, chylomicron formation, and transport into the intestinal lymphatics or portal capillaries. Chylomicrons containing short- and long-chain triglycerides, and the newly incorporated B-100 apoprotein, are preferentially absorbed into the intestinal lymphatics where they are transported into the cisterna chyli, thoracic duct, and systemic circulation where they acquire apolipoproteins C and E from circulating high-density lipoproteins (HDLs). Apolipoprotein (apo) C-II activates lipoprotein lipase in the capillary beds of adipose and skeletal muscle, where they are stored as is or hydrolyzed into free fatty acids, β-monoglyceride, and glycerol. The cholesterol-rich remaining particles (now referred to as chylomicron remnants), return apo C-II molecule to HDL and are recognized by specific hepatic apo E and apo B-100 receptors that rapidly remove them from the circulation by endocytosis. The cholesterol found in chylomicron remnants can be used in very-low-density lipoprotein (VLDL), lipoprotein synthesis, bile acid formation, or cholesteryl storage.
Reverse Transport
HDLs play an important role in the reverse transport of cholesterol from the periphey to the liver. Lecithin cholesterol acyl transferase esterifies HDL cholesterol and cholesteryl esters move to the core of the HDL molecule to allow more free cholesterol to be absorbed into the particle. Continued absorption of free cholesterol and subsequent esterification by lecithin cholesterol acyl transferase leads to the formation of the larger, cholesteryl ester-rich HDL2s. HDL2 molecules continuously acquire cholesteryl esters, resulting in the formation of the HDL1 molecules. On HDL1, cholesteryl esters are transferred from tissues to the liver for disposal or reuse, and not to LDL or VLDL molecules (as in humans), which transfer cholesterol to peripheral tissues. This function of HDL1s may account for the lower incidence of atherosclerotic disorders in dogs compared with humans.23,24
Nucleic Acids
Pyrimidine biosynthesis is one of the classic roles of the liver in nucleic acid metabolism. More recently, microRNAs have been impugned in the normal development and regeneration of the liver, as well as in hepatic pathology. microRNAs are small noncoding RNAs that regulate both messenger RNA and protein expression of target genes, which results in alterations in messenger RNA stability or translation inhibition. microRNAs influence at least one-third of all human transcripts and are known regulators of various important cellular growth and differentiation factors. microRNAs recently emerged as key regulatory molecules in chronic liver disease.25
Metals
The liver incorporates copper into specific copper proteins such as cytochrome c oxidase, mitochondrial monoamine oxidase, and ceruloplasmin. Mobilization of copper from hepatocytes takes place by at least two mechanisms: ceruloplasmin and bile secretion. Cholestatic liver disease is associated with secondary copper retention, which may then induce hepatocyte injury.26,27
Bile Secretion
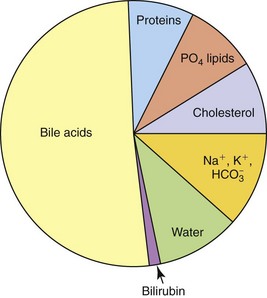
Biliary secretions provide (a) a source of bile acids for fat digestion and absorption, (b) an excretory route for metabolites and xenobiotics, and (c) additional HCO3− for buffering of H+ ion in the duodenum. Bile acids are the major components of bile accounting for about one-half to two-thirds of the total solutes. Bile also contains water, electrolytes, cholesterol, phospholipids, hormones, protein, and bilirubin (Figure 61-2).
Bile components are synthesized, stored, and secreted from the hepatocytes into the biliary ductal system.28 In the absence of neural or hormonal input (as in the fasting state), the gallbladder is relaxed, the terminal biliary ductal sphincter (sphincter of Oddi) is contracted, and bile is largely stored in the gallbladder. While stored in the gallbladder, water and large portions of the electrolytes are reabsorbed by the gallbladder mucosa, concentrating the remaining constituents. During feeding, neural (acetylcholine) and hormonal (cholecystokinin) mechanisms activate gallbladder contraction, biliary ductal sphincter relaxation, and emptying of bile into the duodenum. Secretin and bile salts stimulate bile salt–independent and bile salt–dependent bile flow, respectively.2,5,28
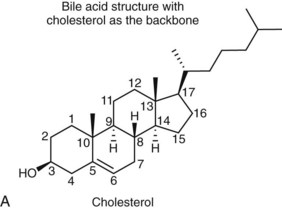
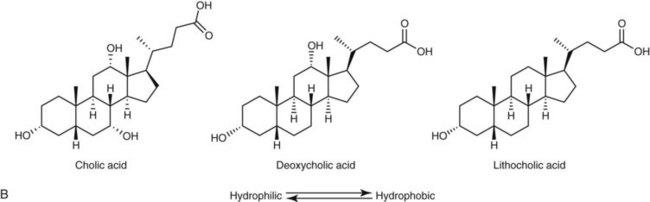
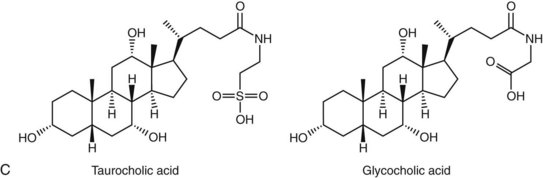
Bile acids are synthesized from the cholesterol nucleus to which are attached a five- or eight-carbon side chain with a terminal carboxylic acid, and hydroxyl groups positioned at the C3, C7, or C12 carbon atom positions (Figure 61-3, A and B). The major primary bile salts are cholic acid and chenodeoxycholic acid in about equal molar quantities. When these primary bile acids are secreted into the lumen of intestine, a portion of each is dehydroxylated by intestinal bacteria to produce the secondary bile acids, deoxycholic acid, and lithocholic acid.2,5,28 Prior to secretion, bile acids are conjugated with taurine and/or glycine to form tauro- and glycoconjugated bile salts (see Figure 61-3, C). Conjugation lowers the pKa to well below the physiologic range of biliary and intestinal pH, and conjugated bile acids become ionized anions (referred to as bile salts) rather than undissociated bile acids. In the ionized form, they are less likely to be absorbed by the small intestine and so maintain a higher intraluminal concentration appropriate for emulsification, digestion, and absorption of lipids. Dogs and cats conjugate primarily with taurine. Dogs can convert to glycine conjugation if taurine is deficient, but cats cannot. Cats are obligate taurine conjugators, and have an essential dietary taurine requirement.2,5,28
Following emulsification and micellarization of fat, most of the secreted bile salts are transported along the GI tract to the ileum where they are absorbed into ileal enterocytes and portal blood flow via Na+–bile salt cotransporters.2,28
Coagulation Factors
The liver plays an important role in maintaining hemostasis. The liver produces procoagulant, anticoagulant, and fibrinolytic proteins, and also removes normal and abnormal clotting factors from the circulation.29
In liver disease, factor and inhibitor synthesis and clearance of activated factors in both the coagulation factors and fibrinolytic system may be impaired. The extent of coagulation abnormalities depends upon the degree of disturbed liver function.29 Patients with hepatic failure may present with the entire spectrum of factor deficiencies and may even develop disseminated intravascular coagulation.
In a study of 42 dogs with histologically confirmed liver disease, one or more coagulation abnormalities were found in 57% of dogs with liver disease.29 Activated partial thromboplastin time was significantly prolonged in dogs with chronic hepatitis with or without cirrhosis. Mean platelet numbers, antithrombin, and factor IX activity were significantly lower in dogs with chronic hepatitis with cirrhosis, compared to dogs with other hepatopathies. D-Dimers were not significantly increased in any group. Only three dogs, all with different histologic diagnoses, satisfied the criteria for disseminated intravascular coagulation. Hemostatic abnormalities were primarily seen in dogs with chronic hepatitis plus cirrhosis, which may be a result of reduced synthesis rather than increased consumption of coagulation factors.
Detoxification
Xenobiotic Agents
Numerous foreign compounds, including drugs, are so hydrophobic that they would remain in the body indefinitely were it not for hepatic biotransformation. Cytochrome P450 (P450 or CYP) comprises a superfamily of enzymes that catalyze oxidation of a variety of xenobiotic chemicals such as drugs, toxic chemicals, and carcinogens, as well as endobiotic chemicals including steroids, fatty acids, prostaglandins, and vitamins. The cytochrome P450 enzymes in families one to three mediate 70% to 80% of all phase I–dependent metabolism of clinically used drugs and participate in the metabolism of a huge number of xenobiotic chemicals. There are 57 known active P450 genes and 58 pseudogenes in the human genome. With 54 active genes, dogs are phylogenetically closest to the human. Although there are many similarities between dogs and humans, there also are many important differences.30–33 Dogs present an interesting challenge in the assessment of P450-mediated drug–drug interactions because most of the enzymes have not been completely characterized, diet and aging induce significant changes in gene expression, and dogs are often treated off-label with a number of human drugs with little idea of risk for drug–drug interaction.
Drug metabolism takes two general forms: phase I metabolism (modification reactions) and phase II metabolism (conjugation reactions). Phase I metabolism typically subjects a drug to oxidation or hydrolysis. It involves the cytochrome P450 (CYP) enzymes, which facilitate reactions that include N-, O-, and S-dealkylation; aromatic, aliphatic, or N-hydroxylation; N-oxidation; sulfoxidation; deamination; and dehalogenation. Phase II metabolism conjugates the drug to hydrophilic substances, such as glucuronic acid, sulfate, glycine, or glutathione. Phase I metabolism usually precedes phase II metabolism, but this is not always the case.34
The toxicity of drugs can be considered in five contexts: on-target toxicity, hypersensitivity and immunologic reactions, off-target pharmacology, bioactivation to reactive intermediates, and idiosyncratic drug reactions.35,36
Ammonia
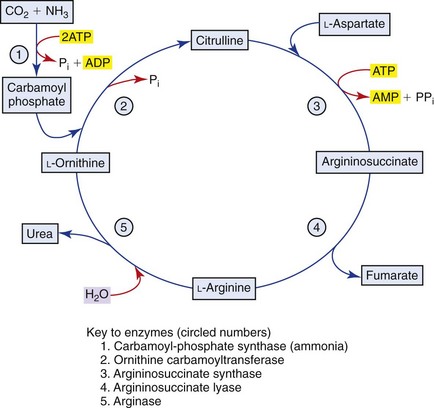
The GI tract, particularly the colon, is the most important source, through the action of bacterial urease on endogenous urea or dietary amines. Ammonia produced by colonic bacteria enters the portal circulation and is transported to the liver for urea cycle transformation (Figure 61-4). Ammonia is transformed into urea in the urea cycle in the overall equation:
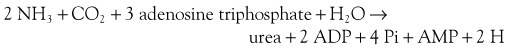
Endogenous Hormones
Mineralocorticoids (aldosterone), glucocorticoids (cortisol, corticosterone), and sex steroids (androgens, estrogens, progesterone) are metabolized by the liver. Changes in the concentrations of total and free cortisol and of the binding capacity of corticosteroid-binding globulin have been reported in canine liver disease. As a consequence of hypercortisolemia, dogs with liver disease and hepatoencephalopathy have clinical and biochemical characteristics of PDH, including polyuria, high basal cortisol levels, and α-melanotropin.37,38 Chronic hypercortisolism is associated with impaired osmoregulation of the release of vasopressin and inadequate urinary concentration.39
Immune Surveillance
The multiple physiologic functions of the liver require an immune response that is locally regulated. Pathogenic microorganisms must be efficiently eliminated, while the large number of antigens derived from the GI tract must be tolerated. The liver favors the induction of tolerance rather than the induction of immunity. Although hepatocytes constitute the major cell population of the liver, direct interaction of hepatocytes with leukocytes in the blood is unlikely. Sinusoidal endothelial cells, which line the hepatic sinusoids and separate hepatocytes from leukocytes in the sinusoidal lumen, and Kupffer cells, the resident macrophage population of the liver, can directly interact with passenger leukocytes. In the liver, clearance of antigen from the blood occurs mainly by sinusoidal endothelial cells through very efficient receptor-mediated endocytosis. Liver sinusoidal endothelial cells constitutively express all molecules necessary for antigen presentation (CD54, CD80, CD86, major histocompatibility complex [MHC] classes I and II, and CD40) and can function as antigen-presenting cells for CD4+ and CD8+ T cells.40,41 Thus, these cells probably contribute to hepatic immune surveillance by activation of effector T cells. Antigen-specific T-cell activation is influenced by the local microenvironment. This microenvironment is characterized by the physiologic presence of bacterial constituents such as endotoxin and by the local release of immunosuppressive mediators such as interleukin-10, prostaglandin E2, and transforming growth factor-β.42
Regeneration
Liver regeneration after partial hepatectomy is a very complex and well-orchestrated phenomenon. It appears to be carried out by the participation of all mature liver cell types.43,44 The process is associated with signaling cascades involving growth factors, cytokines, matrix remodeling, and several feedbacks of stimulation and inhibition of growth related signals.45,46 The liver manages to restore any lost mass and adjust its size to that of the organism, while at the same time providing full support for body homeostasis during the entire regenerative process. In situations when hepatocytes or biliary cells are blocked from regeneration, these cell types can function as facultative stem cells for each other.
Gene expression in the regenerating liver is a multistep process with at least two critical steps: the transition of quiescent hepatocytes into the cell cycle (“priming”), and the progression beyond the restriction point in the G1 phase of the cell cycle. Hepatocytes must first be primed before they can fully respond to growth factors. As many as 70 different genes participate in the early response to hepatectomy, but tumor necrosis factor (TNF), interleukin (IL)-6, and interleukin-22 (IL-22) appear to be the major cytokines involved in the priming of hepatocytes.47 The proliferative effect of TNF on hepatocytes is further influenced by reactive oxygen species, nitric oxide, and glutathione content, and multiple transcription factors (e.g., nuclear factor kappa B, STAT3, AP-1, and C/EBPβ) play major roles in the initiation of early liver regeneration.47 Progression through the cell cycle beyond the initiation phase requires growth factors, primarily hepatocyte growth factor and transforming growth factor-α (TGF-α). The subsequent expression of cell-cycle genes establishes the stage at which replication becomes growth factor-independent and autonomous. At this point, the hepatocyte is irreversibly committed to replicate and the cell cycle replication machinery takes over.
The proliferation of hepatocytes advances from periportal to pericentral ares of the lobules, as a wave of mitoses. Hepatocytes surrounding the central veins are the last ones to undergo cell replication. Proliferation of biliary epithelial cells occurs a little later than hepatocytes. Proliferation of endothelial cells starts at 2 to 3 days and ends around 4 to 5 days after partial hepatectomy. The kinetics of proliferation of stellate cells is incompletely understood. The regenerative capacity of the residual hepatocytes may restore liver mass and function after as much as 65% to 70% hepatectomy and it takes place over 7 to 14 days in most animal species.48 A small wave of apoptosis in hepatocytes occurs at the end of regeneration.
History and Physical Examination
Hein P. Meyer and Jan Rothuizen
Clinical Importance
The liver is the second largest organ in the body and performs an estimated 1500 essential biochemical functions.1 These diverse functions include drug metabolism; removal of exogenous and endogenous toxins (e.g., ammonia, food antigens); synthesis of vital substances such as albumin and blood clotting factors; protein, fat, and carbohydrate metabolism; vitamin storage and activation; glycogen, triglyceride, and mineral (e.g., copper, iron) storage; activation, conversion, secretion, deactivation, and excretion of various hormones; bile salt synthesis; conjugation and excretion of bilirubin in bile; among others. Symptoms (defined here as abnormalities noted by the owner), clinical signs (defined here as abnormalities found during the physical examination), and diagnostic results reflect impairments in these functions. Hepatitis represented approximately 1% of the clinical population of the companion animal teaching hospital of Utrecht University. Box 61-1 summarizes the most common liver diseases with their possible etiologies in dogs and cats.
Box 61-1
Causes of Acute Liver Disease in Dogs and Cats
Drugs and Anesthetics
Herbal and Dietary Supplements
History of Dogs and Cats with Liver Disease
A properly taken history is pivotal to defining the most clinically relevant problems that need to be resolved. A structured interview process and understanding the basics of communication are important success factors to retrieve this crucial information. Fortunately, the knowledge about communication in the medical profession and the focus on the veterinary curriculum, has increased considerably during the last few years.2,3
Some basic principles should be kept in mind to understand the symptoms in dogs and cats with diseases affecting the hepatic parenchyma, portal vasculature, and the biliary system. First, for most of its functions, the liver has a tremendous (approximately 80%) reserve capacity and a remarkable potential to regenerate.4 Symptoms occur only when progressive disease exhausts hepatic reserves. Diseases often remain subclinical for lengthy periods of time; symptoms may be relatively mild and nonspecific because the liver reserve prevents overt abnormalities. Symptoms such as lethargy, vomiting, or mild polyuria and polydipsia (PU/PD) may alert the clinician that a liver disorder could be developing. Serious symptoms may indicate loss of hepatic reserves. The onset of symptoms may be acute, but they may be the end result of a disease that has been present for many weeks or months. Because no specific physical abnormalities occur with most liver diseases, it is important to remember that liver disease may be present when symptoms of illness are unexplained or nonspecific. Sensitive and specific laboratory tests may easily detect such liver diseases.5
Second, most liver diseases cause similar signs and symptoms (Table 61-2). One of these is acholic feces, which occurs nearly exclusively in dogs with common bile duct obstruction.6 Owners may note the light-gray appearance of stool, which in combination with icterus is virtually diagnostic for extrahepatic cholestasis. Different combinations of clinical signs and symptoms may occur with any liver disease. Statistically, one disease may be associated with a typical pattern of signs and symptoms in dogs and cats. However, overlapping patterns are so great that it is useless to try to identify the exact disease based on clinical signs and symptoms alone (Table 61-2). Clinical signs and symptoms associated with liver diseases of cats are similar to those in dogs, except for PU/PD, which is not clinically overt in most cats. Certain liver diseases cause neurobehavioral signs associated with hepatic encephalopathy,7 and these signs may wax and wane in their frequency and severity. Any medication given may appear to be effective because of the natural fluctuation of signs. Therefore signs of hepatic origin may be easily missed. Seizures alone are never caused by hepatic encephalopathy; if they do occur, they occur in combination with other signs seen with this syndrome.5 Furthermore, very few, if any, medications to treat liver diseases have been tested in double-blind, placebo-controlled studies, making decisions about the best therapeutic regimens difficult.8
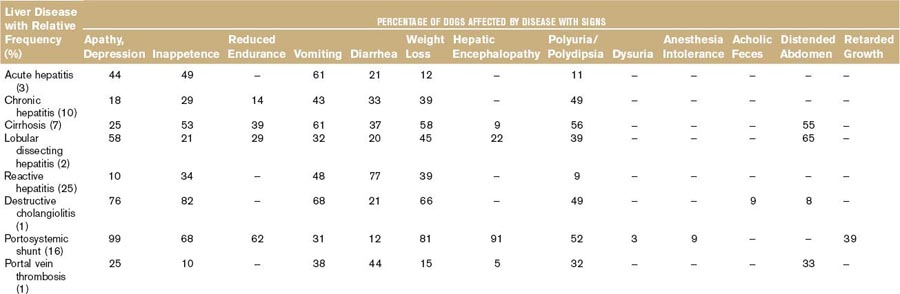
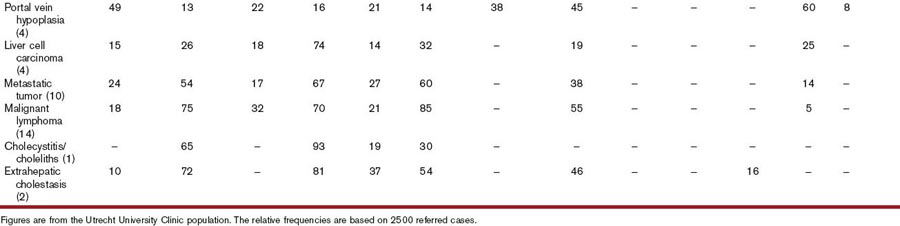
It is usually not possible to differentiate between hepatic diseases and diseases of other organs based on symptoms and clinical signs. Signs associated with hepatic diseases are nonspecific; similar signs may occur in diseases of many other organ systems, most notably the GI, neurologic, renal, and hematologic systems (see Table 61-2).9 Of the GI tract–related symptoms, nausea expressed as vomiting in acute cases or reduced, irregular appetite with occasional vomiting and weight loss over time, is very common in liver and biliary diseases of dogs and cats. For biliary diseases these are always the most prominent symptoms. Diarrhea, however, is not a major symptom of liver disease, and in cases in which diarrhea is the leading symptom the liver is only rarely the causative organ (except for rare cases with complete common bile duct obstruction). A rare sign (not included in Table 61-2) is an ulcerative form of dermatosis, so-called superficial necrolytic dermatitis, hepatodermal, or hepatocutaneous syndrome. This syndrome occurs rarely in dogs with liver cirrhosis and nodular hyperplasia and whose pathogenesis is poorly understood.10 This symptom and the more common symptoms of lethargy, inappetence, vomiting, diarrhea, weight loss, PU/PD, and neurobehavioral symptoms are frequently associated with diseases of other organs. Therefore the history often discloses symptoms that may suggest hepatic disease, but may also be caused by other disorders.
Two main reasons account for the nonspecificity of liver-related symptoms. First, the liver is the central organ for many metabolic and detoxifying pathways; consequently, failing liver function may cause dysfunction of other organs. One example is hepatic encephalopathy; metabolic dysfunctions of the liver cause neurotransmitter dysfunctions of the brain, resulting in neurobehavioral signs.11 Second, toxic factors resulting from diseases of other organ systems (especially from the GI tract) often secondarily affect the liver. Examples include hepatic lipidosis in diabetes mellitus, steroid-induced hepatopathy in Cushing syndrome, reactive hepatitis in GI diseases, and centrolobular liver necrosis in acute, severe anemia.5 Therefore signs and symptoms of liver disease may be hidden within signs of other organ dysfunction, and vice versa. Because clinical and physical examination findings may be compatible with hepatic disease, and because laboratory tests to detect hepatic disease are also abnormal with primary and secondary hepatopathies, it is often necessary to make a histologic diagnosis of the liver disorder to resolve this dilemma.12,13
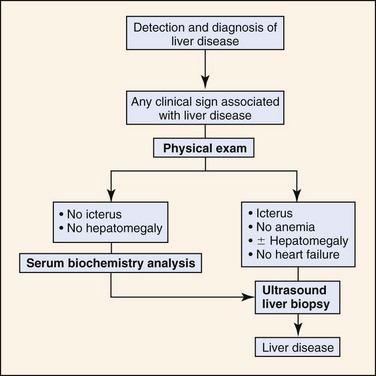
Lack of specific physical examination findings may prevent recognition of a primary liver disease. Most dogs with illnesses causing the clinical signs listed in Table 61-2 are candidates for having a primary hepatopathy. In all such cases, further diagnostic studies should be performed to confirm or exclude liver disease (Figure 61-5).
Predispositions
Breed, sex, age, and drugs may predispose dogs and cats to hepatopathies. The presence of numerous risk factors should be a stimulus for an extended diagnostic workup; other diseases should be investigated in the absence of such suspicions. Caused by hypersensitivity to sulfonamides, destructive cholangiolitis is the most common drug-induced liver disease.14 A recent history of therapy with sulfonamides or other potentially hepatotoxic drugs, combined with icterus makes this condition likely and should prompt immediate discontinuation of the medication. Breed associations may occur when a disease is (in part) determined by genetic factors. Breeders may, by chance, increase the incidence of hepatic diseases by familial selection. Because dog breeds may represent more or less closed populations in a country, breed predispositions may vary among countries. Therefore this section only mentions generally applicable predispositions; locally, other breed associations may be more pertinent.
Chronic hepatitis and cirrhosis, both of which are, as a rule, different stages of one disease, occur more frequently in certain breeds.15 Hepatitis may develop at any age, but typically not before 2 years of age. Only lobular dissecting hepatitis tends to occur at a young age (i.e., often before 1 year of age).16 Breeds associated with hepatitis are Doberman Pinschers, Bedlington Terriers, West Highland White Terriers, American and English Cocker Spaniels, Labrador Retrievers, and many other breeds. Recent copper excretion studies have shown that hepatitis is caused by copper retention and not vice versa in Doberman Pinschers.17 The cause of copper retention remains unclear; many of the tested candidate genes (including Murr 1, the affected gene in Bedlington Terriers) were excluded as monogenetic causes for copper-associated subclinical hepatitis in Doberman Pinschers.17 The hepatitis in Doberman Pinschers is sex linked, confined to females, and aggressive.18 It is responsive to medication with penicillamine,19 but may terminate in micronodular cirrhosis. This form of cirrhosis, predominantly seen in copper toxicosis, differs from other forms of chronic hepatitis, in which patients typically develop macronodular cirrhosis with large hyperplastic nodules. Hepatitis is overrepresented in female Doberman Pinschers by a factor of 10; a study in Finland showed that approximately 10% of Doberman Pinschers may be affected.20 Inherited copper toxicosis is also a well described entity in Bedlington Terriers worldwide.21 Both sexes may be affected. Clinical signs usually develop after 4 years of age as a result of the gradual accumulation of copper. It is caused by a defect in the Murr 1 gene, leading to a severely decreased excretion of copper by hepatocytes. Other affected breeds are West Highland White Terriers (particularly in the United States), Skye Terriers, Dalmatians, Anatolian Shepherd dogs, and Labrador Retrievers.22–26 Siamese cats may also be predisposed to copper-associated hepatopathies.9 Although essential for life, copper is usually ingested to excess and must be eliminated by the liver to prevent toxicity. The central role of the liver in copper homeostasis makes it vulnerable if elimination processes fail.27 Furthermore, increased copper levels add to the oxidative stress, which is an important component in chronic inflammatory and cholestatic diseases in dogs.25 Spaniels seem to have the form of chronic hepatitis unrelated to copper toxicosis and develop macronodular cirrhosis when left untreated. No sex predisposition exists, but there seems to be a worldwide overrepresentation of hepatitis in this breed.
Congenital portosystemic shunts (CPSS) are seen in both sexes in various breeds. Intrahepatic shunts predominate in large breeds, whereas extrahepatic shunts predominate in small and toy breeds. Although CPSS are most likely inherited in some fashion in all affected breeds, this has only been proven in Irish Wolfhounds28 and Cairn Terriers.29 Worldwide predispositions occur in Irish Wolfhounds, Australian cattle dogs, Labrador Retrievers, Dachshunds, Yorkshire Terriers, Cairn Terriers, Maltese Terriers, and Miniature Schnauzers.7,9 In the United States, an increased prevalence of shunts has also been reported to occur in German Shepherd dogs, Doberman Pinschers, and Golden Retrievers. CPSS occur most often in mixed-breed cats; however, Persian and Himalayan cats are frequently overrepresented. Clinical signs are usually seen in young dogs and cats (<1 year old) with congenital shunts.5,9
Pathogenesis of Common Symptoms of Primary Liver Diseases
Vomiting
Vomiting is one of the most common symptoms noted in dogs and cats with liver disease. Vomiting may be caused by direct stimulation of the vomiting center via the chemoreceptor trigger zone in the fourth ventricle by (endo)toxins that are not cleared by the liver.30 This typically occurs when toxins from the GI system bypass the liver and access other body systems. Vomiting is common in all conditions that share portosystemic shunting and liver dysfunction (e.g., congenital shunts and acquired shunts because of hepatitis, fibrosis, cirrhosis, and portal vein hypoplasia or thrombosis). Hepatic diseases that cause an abnormal liver shape may reposition the upper GI tract and induce nausea and vomiting by vagal stimulation. Causes include hepatic tumors, especially liver cell (or hepatocellular) carcinomas, and unilateral collapse and contralateral hypertrophy, which may occur with thrombosis of a main branch of the portal vein. The gallbladder and larger bile ducts have a rich sympathetic innervation; therefore dilation (e.g., extrahepatic cholestasis), cholecystitis, or cholelithiasis should be suspected in vomiting dogs and cats.5
Vomiting is also common in upper GI disease. In many GI diseases, translocation of bacteria and endotoxins may cause secondary, nonspecific, reactive hepatitis.30 This occurs frequently in dogs, but rarely in cats. Reactive hepatitis is characterized by intrahepatic canalicular cholestasis, liver cell necrosis, and an exudative inflammatory reaction. Clinical signs, symptoms, and diagnostic results associated with primary liver disease and reactive hepatitis are similar; therefore, a further diagnostic workup is important to reveal the primary cause.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


