30 Hyperlipemia
Hyperlipemia is a life-threatening medical condition seen almost exclusively in pony, donkey and miniature horse breeds. It results from sudden and severe breakdown of body fat stores and nutrition plays a key role in the pathogenesis and treatment of the condition. Intensive therapeutic intervention is an important component in achieving a successful outcome although an overall survival rate of only 56% is reported in six case series published since 1992 (Durham 2006, Hallebeek & Beynen 2001, Mogg & Palmer 1995, Rush-Moore et al 1994, Waitt & Cebra 2009, Watson et al 1992b).
Epidemiology and risk factors
Risk factors for hyperlipemia may be considered in two categories: pre-existing inherent factors that predispose an individual to hyperlipemia; and then precipitating factors that trigger hyperlipemia in predisposed individuals (Table 30-1).
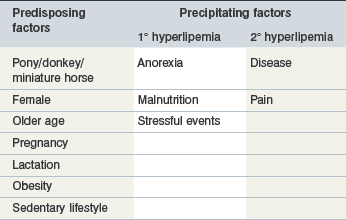
Predisposing factors
Predisposition to hyperlipemia is largely related to insulin resistance (IR) and increased nutritional requirements. Pony, donkey and miniature horse breeds demonstrate a relative insensitivity to insulin that may be further compounded by obesity and a sedentary lifestyle (Jeffcott et al 1986, Reid & Mohammed 1996). IR facilitates mobilization of fat and glycogen stores whenever caloric intake is insufficient (Jeffcott et al 1986, June et al 1992) and thus represents an inherent prolipolytic status that predisposes to hyperlipemia (Breidenbach et al 1999). Mature animals appear predisposed to hyperlipemia (Jeffcott & Field 1985, Reid and Mohammed 1996) perhaps as a result of decreasing insulin sensitivity (Murphy et al 1997) although cases are occasionally encountered in foals and even neonates (Gilbert 1986, Hughes et al 2002, Tan et al 2005). Pregnancy is also associated with IR (Fowden et al 1984) and, moreover, increased nutrient demands that further promote fat mobilization. The latter is also the likely explanation for increased hyperlipemia risk associated with lactation.
Females appear predisposed to hyperlipemia largely due to pregnancy or lactation, although one study found female donkeys to be at twice the risk of males even when not pregnant or lactating (Reid & Mohammed 1996). Examination of data from 135 published cases of hyperlipemia shows 1.5 times as many reproductively inactive females compared with males also supporting that being female is a risk factor independently from pregnancy and lactation (Fig. 30.1). Further unpublished data reveals a significantly higher serum insulin concentration in female than male donkeys supporting IR as a factor in this gender predisposition (N. du Toit, personal communication).
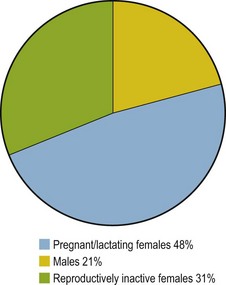
Figure 30.1 Distribution of hyperlipemia cases according to gender and reproductive status. Data derived from 135 published cases (Durham 2006, Gay et al 1978, Jeffcott & Field 1985, Mogg & Palmer 1995, Naylor et al 1980, Oikawa et al 2006, Rush-Moore et al 1994, Waitt et al 2009, Watson et al 1992b).
Precipitating factors
Factors known to trigger hyperlipemia in predisposed individuals comprise inadequate feed intake, stress (e.g., feed changes, transportation), pain and disease. The neuroendocrine response to these factors includes increased adrenocorticotrophic hormone, antidiuretic hormone, growth hormone, catecholamines, glucocorticoids and glucagon and suppression of insulin (de Pew et al 1994, Liddell et al 1979, Sticker et al 1995, Stoner 1987). This endocrine milieu favors lipolysis and may trigger hyperlipemia in sick and/or hypophagic subjects.
Primary hyperlipemia occurs in the absence of a precipitating disease process whereas secondary hyperlipemia results from a separate inciting disease, such as enterocolitis, parasitism, colic, and impactions (Mogg & Palmer 1995, Rush-Moore et al 1994, Schotman & Wagenaar 1969, Watson et al 1992b, Waitt & Cebra 2009).
Pathophysiology
Processes controlling fat mobilization
The delivery of fatty acids (FAs) from stored triacylglycerols (TGs) in adipose tissue for oxidation by other cells represents the major energy supply route through the body, especially at times of negative energy balance. This mobilization is achieved by three intercoordinated processes (Fig. 30.2):
1. In adipose tissue: stored TG is hydrolyzed to release free FAs and glycerol into the plasma.
2. In the liver: FAs and glycerol are re-esterified into TGs and exported within very low density lipoprotein particles (VLDLs).
3. In tissues throughout the body (e.g. muscle): TG in VLDLs is hydrolyzed to release FAs which are then absorbed and oxidized as an energy source.
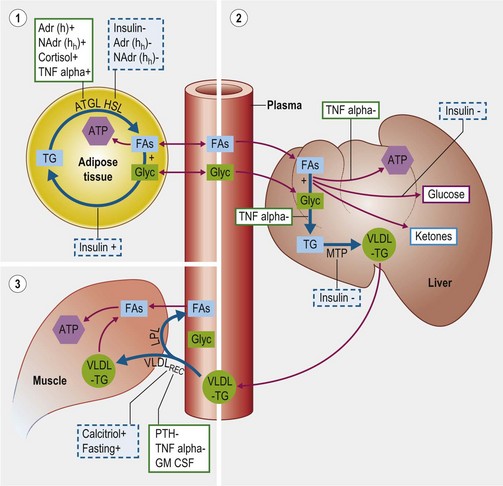
Figure 30.2 Schematic diagram of physiologic and pathophysiologic mechanisms controlling fatty acid and lipid transport.
These three processes are described in further detail below.
Adipose lipolysis
Mechanisms (Fig. 30.2, section 1)
The FAs and glycerol stored in adipose TG are released by the coordinated actions of three enzymes: adipose triglyceride lipase (ATGL), hormone sensitive lipase (HSL), and monoacylglycerol lipase (Vaughan et al 1964, Villena et al 2004, Zechner et al 2009). Following their release, FAs and glycerol may be used for energy by the adipocyte or enter the plasma where they are primarily transported to the liver.
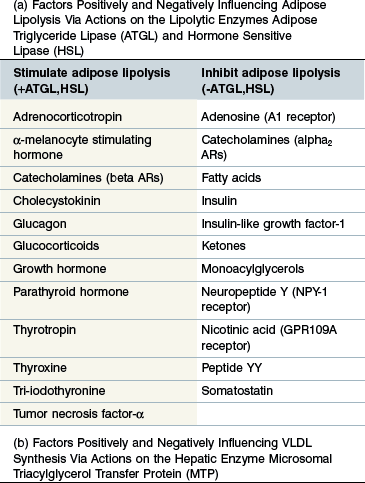
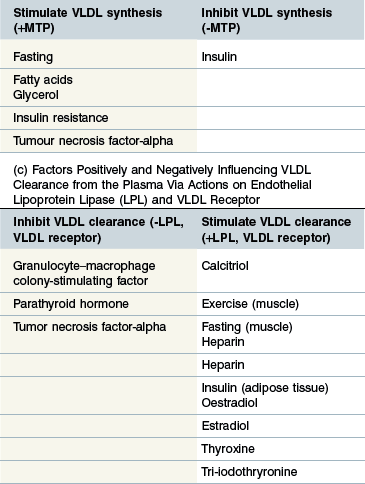
Regulation (Table 30-2a)
ATGL and HSL represent the key, rate-limiting steps in adipose TG hydrolysis and are both closely controlled by the endocrine system. Both ATGL and HSL lipolytic activity is stimulated by catecholamines (beta adrenoreceptors) and inhibited by insulin (Large & Arner 1998). Additional regulatory factors are listed in Table 30-2a (Coppack et al 1994, Large & Arner 1998, Zechner et al 2009). Insulin may further limit FA release from adipose stores by promoting re-esterification of free FAs and glycerol into TG. Regulation of the two enzymes differs slightly in that HSL activity increases during prolonged fasting whereas ATGL is acutely up-regulated following feed withdrawal, probably in response to increased glucocorticoid levels (Villena et al 2004, Zechner et al 2009). Tumor necrosis factor-α (TNF-α) is a potent stimulator of lipolysis during sepsis probably via stimulation of ATGL (Zechner et al 2009).
Hepatic VLDL synthesis and export
Regulation (Table 30-2b)
The rate limiting step in hepatic VLDL synthesis is controlled by microsomal triacylglycerol transfer protein (MTP) (Hussain et al 2003). Insulin is a potent inhibitor of MTP and therefore suppresses VLDL synthesis (Kamagate et al 2008). Consistent with this it has been shown that MTP (and therefore hepatic VLDL synthesis) is significantly upregulated during fasting and also in IR states (Bartels et al 2002, Kamagate et al 2008). Increased substrate availability (free FAs and glycerol) following adipose lipolysis induced by fasting is another factor promoting VLDL synthesis (Lewis et al 1995). TNF-α has also been shown to increase hepatic TG synthesis by stimulating FA synthesis by the liver and inhibiting oxidation of FAs thereby increasing substrate for TG synthesis (Chen et al 2009).
Extraction of FAs from plasma VLDL-TG
Mechanisms (Fig. 30.2, section 3)
In peripheral capillary beds (e.g. muscle, fat), VLDL interacts with endothelial lipoprotein lipase (LPL) which releases FAs and glycerol from the TG within. The free FAs enter cells locally to be oxidized as an energy source (or re-esterified in adipose tissue) while most glycerol returns to the liver for further metabolism. An additional mechanism for peripheral VLDL clearance involves the endothelial VLDL receptor which internalizes VLDL particles in their entirety (Takahashi et al 2004).
Regulation (Table 30-2c)
Most LPL exists in adipose tissue and this is stimulated by insulin whereas muscle LPL is upregulated during fasting and exercise (Eckel 1989, Kern 1997, Knapper et al 1995). Heparin and calcitriol both upregulate LPL expression whereas TNF-alpha and parathyroid hormone (PTH) have an inhibitory effect (Chen et al 2009, Querfeld et al 1999, Tornvall et al 1995). It has been suggested in the past that azotemia inhibits LPL in horses although there is no good evidence in support of this (Naylor et al 1980, Vaziri 2009). Regulation of the VLDL receptor appears broadly similar to that of LPL but is also upregulated by thyroid hormones and estradiol and downregulated by granulocyte–macrophage colony-stimulating factor (GM-CSF) (Ishibashi et al 1994, Jokinen et al 1994, Kwok et al 1997, Masuzaki et al 1994).
Key Points
• The flux of FAs and glycerol from adipose stores, via the liver to peripheral tissues, is a key energy supply route and comprises several highly regulated, balanced, interdependent and coordinated enzymatic processes
• FAs and glycerol are released into the plasma from TGs stored in adipose tissue
• Plasma FAs and glycerol are re-esterified back to TG by the liver and secreted into the plasma within VLDL particles
• The TG within plasma VLDL particles is hydrolyzed back to FAs and glycerol in peripheral tissues for use as an energy source
Pathophysiology of hyperlipemia
Hyperlipemia inevitably follows when adipose lipolysis (ATGL- and HSL-dependent) and consequent hepatic VLDL synthesis (MTP- and substrate-dependent) exceeds the rate of clearance of plasma VLDL (LPL- and VLDL receptor-dependent). Risk factors for hyperlipemia (Table 30-1) are likely to be associated with conditions including IR, negative energy balance, increased sympathetic and adrenocortical activity and systemic inflammation. Under such conditions it would be expected that adipose lipolysis and hepatic VLDL synthesis would be increased (Table 30-2a,b, Fig. 30.2). Anorexia increases VLDL clearance by muscle (but not fat), although this process could be antagonized by inflammatory cytokines (Fig. 30.2, Table 30-2c). In a study of hyperlipemic ponies, Watson et al (1992a) found increases in the rates of all three key processes: adipose lipolysis, hepatic VLDL synthesis, and VLDL clearance. However, the majority of the cases studied were primary hyperlipemia and it is possible that effects on VLDL clearance could be different in hyperlipemia secondary to inflammatory diseases due to the inhibitory effects of cytokines (TNF-α and GM-CSF) on LPL and the VLDL receptor (Table 30-2c).
Clinical signs
Many differing clinical signs occur with hyperlipemia (Box 30.1) but inappetance, dullness, depression and weakness are almost invariable. Any clinical signs present might reflect a cause and/or effect of hyperlipemia and this represents a significant diagnostic challenge. When clinical signs result from hyperlipemia per se, they are probably a result of poor peripheral perfusion and fatty infiltration of tissues and organs that may result in multiorgan dysfunction, most frequently affecting the liver and kidneys.



