Chapter 17 Hepatoencephalopathy
Pathophysiology and Mechanisms
Pathophysiology
HE is a complex pathophysiologic state that is probably multifactorial in origin.1 It is generally accepted that gut-derived substances of bacterial and protein metabolism are important in the pathogenesis. Evidence for this includes the observation that reduction of gut bacterial flora or dietary protein often result in improvement of neurologic function without altering underlying hepatic disease.
Although ammonia has long been implicated as being the most likely neurotoxin involved in HE, actual mechanisms are not well understood despite decades of research. Evidence is accumulating that brain glutamatergic system and astrocytic (glial) function is significantly deranged in HE as a result of their role in detoxifying ammonia.2,3 Other theories proposed in the past (e.g., alteration in monoamine or catecholamine neurotransmitters because of perturbed aromatic amino acid metabolism, alteration in amino acid neurotransmitters, increased brain γ-aminobutyric acid [GABA], increased cerebral levels of an endogenous benzodiazepine-like substance) have little support today.
There are several observations that provide support for a central role for ammonia in the pathogenesis of HE.4 Encephalopathy can be precipitated in cirrhotic patients by ingestion of ammonia-generating substances (e.g., protein, urea, ammonium salts). Congenital hyperammonemia caused by urea cycle disorders causes HE and coma in children and dogs. In addition, therapy decreasing intestinal production and absorption of ammonia (e.g., low-protein diet, lactulose administration, reduction of gut bacteria by antibiotics) usually results in improvement of clinical signs. Also of interest is the observation that metabolic derangements that increase movement of ammonia across cell membranes or increase ammonia production can precipitate HE in susceptible patients. Such derangements include alkalosis (which facilitates brain uptake of ammonia by increasing the concentration of ammonia base) and profound hypokalemia (which potentiates alkalosis and increases renal ammonia production).
That glutamate plays a central role in HE is suggested by the finding that there are many alterations of brain glutamate in various models of HE, and virtually all aspects of the glutamate system are altered (e.g., synthesis, metabolism intercellular trafficking, function and expression of glutamate transporters and receptors).2 Many of these functions occur in astrocytes (glial cells), cells that are consistently altered histopathologically in HE.
Evaluation of the Patient
History
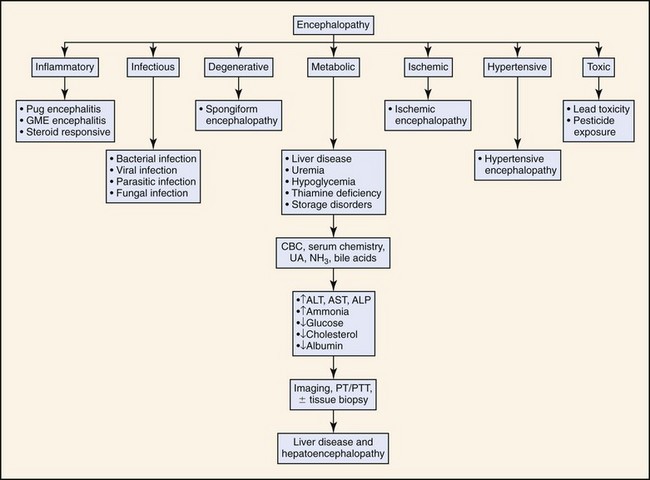
Clinical presentation of cats with HE is similar to dogs but has some unique aspects. Hypersalivation is the most frequently reported clinical abnormality in cats with HE but is rarely reported in dogs. Seizures also appear to occur more frequently in cats than dogs with HE. Seizures occurred in approximately 50% of feline cases reported in the literature. In contrast, seizures are not a particularly common feature of canine HE. Inappropriate aggression is also relatively frequently in cats compared to dogs. In contrast, compulsive behavior (head pressing, circling, aimless wandering) is observed more frequently in dogs than cats. Neurologic signs such as disorientation, ataxia, and stupor are frequently observed in both species. Figure 17-1 outlines the diagnostic approach to patients with signs of encephalopathy. Gastrointestinal abnormalities such as vomiting, diarrhea, and anorexia are reported less frequently in cats than dogs. Polyuria and polydipsia are also less frequently reported in cats than dogs. Risk factors that may precipitate HE in susceptible patients include alkalosis, hypokalemia (which may result from profuse vomiting or diarrhea or from excessive diuretic therapy with furosemide or other loop diuretics), anesthetics and sedatives (attributed to both increased cerebral sensitivity to the drugs and to impaired drug elimination; benzodiazepines are relatively safe), gastrointestinal hemorrhage (a common precipitating cause of HE is gastroduodenal ulceration which is common in patients with hepatic disease), transfusion of stored blood (it may contain high concentrations of ammonia), constipation (it increases colonic absorption of neurotoxic products of bacterial protein digestion), and methionine administration.
< div class='tao-gold-member'>
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


