Chapter 4 Gastrointestinal Inflammation
The fundamental biology of the acute and chronic inflammatory responses is well described in numerous texts.1,2 Acute inflammation occurs within hours to days of exposure to an inciting trigger and is characterized by changes such as vasodilation, tissue edema and protein exudation, neutrophilic exocytosis, and the release of a wide range of cell-derived preformed or newly synthesized inflammatory mediators. By contrast, the chronic inflammatory response generally occurs subsequent to this acute reaction (over weeks to months) or may have a distinct inciting trigger. Chronic inflammation involves tissue infiltration by mononuclear inflammatory cells (macrophages with formation of multinucleate giant cells, lymphocytes, and plasma cells) with production of a separate range of proinflammatory mediators, cytokines and chemokines. The chronic inflammatory response may incorporate aspects of tissue necrosis and remodeling involving stromal growth factors, matrix metalloproteinases and their inhibitors, and progress to the formation of granulation tissue and organized fibrosis.
The challenge of interpretation of alimentary inflammation is compounded by the intrinsic relationship between the immune and inflammatory responses in these mucosal tissues. The gastrointestinal mucosae are normally populated by large numbers of leukocytes involved in the provision of innate and adaptive immune surveillance.11,12,15,51 These cells may be diffusely present throughout the lamina propria or marshalled into organized lymphoid structures and their function is described elsewhere in Chapter 3. These immune populations are designed to respond to the plethora of antigenic substances that naturally pass over the mucosal surface, although the outcome of such responses may be immunologic tolerance rather than activation of protective immunity. Many of these lymphoid responses are therefore regarded as physiologic rather than pathologic, so a major complication in interpreting gastrointestinal inflammation is distinguishing between cellular proliferation occurring to maintain homeostasis versus proliferation indicating a true inflammatory reaction.
It remains the case that our assessment of alimentary inflammation is largely made by subjective evaluation of a hematoxylin and eosin (H&E)-stained section of biopsy tissue. As is discussed in Chapter 29, this observational process is fraught with potential pitfalls, largely relating to lack of standardized criteria, which define changes consistent with inflammation and distinguish these from baseline physiological infiltration. To that end, the World Small Animal Veterinary Association (WSAVA) Gastrointestinal Standardization Group recently proposed histologic guidelines by which gastrointestinal inflammatory responses may be interpreted in a more standardized fashion.10
In the research laboratory, it has become possible to more rigorously define alimentary inflammatory processes by the application of immunohistochemical phenotyping of mucosal leukocyte subpopulations and enumeration of these per unit area of mucosa. Flow cytometric analysis of disaggregated mucosal biopsies has also been employed for the phenotypic characterization of intraepithelial and lamina propria leukocytes.45,55 Additionally, immunohistochemical, biochemical or molecular tools have been used to characterize the local tissue production of proinflammatory mediators including cytokines, chemokines, and matrix metalloproteinases. Preliminary studies now report the role of “pattern recognition receptors” (PRRs) expressed by antigen-presenting cells (APCs) in the initial interaction between pathogens and the alimentary immune system. For example, cultured canine colonic epithelial cells express both membrane (Toll-like receptors 2 and 4) and intracytoplasmic (nucleotide-binding oligomerization domain [NOD]2) PRRs, and when stimulated by appropriate ligands (lipopolysaccharide or peptidoglycan) display upregulation of genes encoding the cytokines interleukin (IL)-7 and IL-8.50 These PRRs are also naturally expressed within the canine intestinal mucosa.24,28 Despite these advances, there remains much to learn about canine and feline alimentary inflammation. For example, the interaction between an array of neuropeptides and the intestinal immune and inflammatory processes remains to be investigated. Dogs and cats appear to lack Paneth cells of the basal crypt region of the intestine that in other species have a major role in the production of assorted bacteriocidal toxins (lysozyme, defensins) and may act to protect the adjacent crypt epithelia. Whether these species have an alternative source of these key inflammatory mediators is also unknown. The application of immunohistochemical and molecular techniques to the study of canine and feline gastrointestinal inflammatory disease are described using selected clinical examples in the sections that follow.
Oral Inflammation
One of the most severe and clinically challenging examples of oral inflammatory disease is feline chronic gingivostomatitis. This disorder likely has multifactorial etiopathogenesis involving dental disease, feline calicivirus infection, oral bacterial infection, and the exuberant mucosal immune response to assorted antigenic materials. The histopathologic appearance is of a severe, predominantly plasmacytic inflammatory reaction with the presence of distinctive large plasma cells (Mott cells) containing an accumulation of immunoglobulin protein as cytoplasmic Russell bodies (Fig. 4-1). Other cell types may also be part of this mucosal infiltrate, particularly lymphocytes and eosinophils.
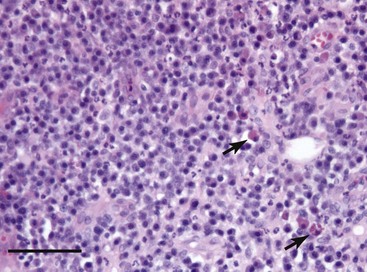
Recent immunohistochemical investigations have characterized the normal baseline resident leukocytes in the feline oral mucosa20 and defined how these alter in this chronic inflammatory process. The mucosal infiltrates in cats with chronic gingivostomatitis are dominated by immunoglobulin (Ig) G plasma cells and recently emigrated myelomonocytic cells expressing the marker MAC387. There are also appreciable numbers of CD3+ T lymphocytes, and of these, CD8+ cells dominate over CD4+ T cells with a median CD4-to-CD8 ratio of 0.22. Surprisingly few IgM or IgA plasma cells, and sparse mast cells, are present in these lesions. APCs expressing class II molecules of the major histocompatibility complex (MHC) are also present, principally forming a subepithelial and perivascular network.19 This reaction has been functionally categorized as a mixed T-helper 1 (Th1) and Th2 immune response by analysis of tissue cytokine gene expression,22 and affected cats are also known to have alterations in salivary immunoglobulin concentrations (elevated IgG and IgM with decreased IgA).21
Gastric Inflammation
The etiopathogenesis of gastritis has been relatively poorly studied in small companion animals. It is clear that the gastric mucosa has a normal complement of resident leukocytes that maintain immune surveillance and that “gastric lymphoid aggregates” within the lamina propria are a normal feature of the histology of this organ. The follicular microarchitecture and immunophenotype of lymphoid cells within these aggregates has been characterized in the normal dog.25 As is discussed in Chapter 56, gastritis may be primary in nature, or may occur in association with an inflammatory response in the intestinal tract. Gastritis may have a defined etiology identified from history or clinical examination (e.g., irritant gastritis) or may be classed as idiopathic in nature.
The specific characterization of gastric inflammation relies on examination of biopsy tissue, which is most often collected endoscopically. A thorough histologic examination of the gastric mucosa necessitates collection of biopsy samples from both the fundic and antral-pyloric regions, as lesional change may be recognized in either or both of these sites. The distinct microarchitecture of these anatomical locations means that histologic patterns of inflammatory change may differ, and for this reason the WSAVA Gastrointestinal Standardization Group (see previous discussion) defined specific criteria related to the assessment of inflammatory change in the gastric fundus and antrum.10
All forms of acute and chronic inflammatory change may be recognized within the gastric mucosa, although lymphoplasmacytic or mixed lymphoplasmacytic and eosinophilic inflammation is probably more commonly noted than neutrophilic, granulomatous, pyogranulomatous, or pure eosinophilic gastritis.49 Leukocyte infiltration of the lamina propria may be accompanied by microarchitectural disturbances involving the surface epithelium (subtle degenerative change through to ulceration), deep epithelium (degenerative change within the fundus or hyperplasia within the antrum) or glandular tissue (“mucosal atrophy” characterized by fibrosis or “glandular nesting”).
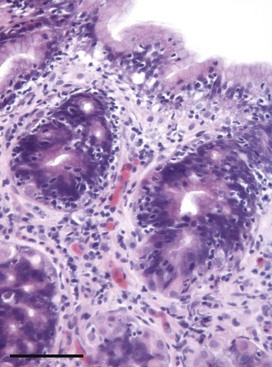
Lymphoplasmacytic inflammation is characterized by infiltration of these cell types into the lamina propria and is often accompanied by distinct increases in the number of intraepithelial lymphocytes (IELs) within both surface and deep epithelial structures (Fig. 4-2). In the fundic mucosa, lymphocytes also often infiltrate and apparently disrupt the glandular tissue. It is likely that the majority of these epitheliotropic lymphocytes are CD3+ T cells and probably CD8+ cytotoxic cells, although immunohistochemical characterization has not been widely reported. In humans, this histologic pattern characterizes the autoimmune disease “atrophic gastritis,” and although the similarity has been noted, there is no proposal at this time that the canine or feline reaction pattern represents an equivalent disorder. Another distinctive feature of gastric inflammation (particularly lymphoplasmacytic gastritis) in dogs and cats is hyperplasia of the gastric lymphoid aggregates, which may sometimes be extreme and occupy up to 50% of the tissue area of any one biopsy.35
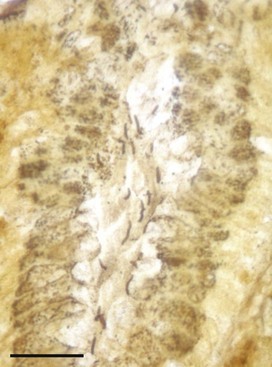
One of the most contentious areas in small animal gastroenterology is the debate concerning the role of Helicobacter spp. in the induction of gastritis.37 These distinctive bacteria are frequently noted associated with the surface epithelium and gastric pits of canine and feline tissue biopsies with entirely normal microarchitecture (Fig. 4-3); however, there have been clinical and experimental research studies that have proposed them as an etiologic factor in gastric mucosal inflammation and lymphoid aggregate hyperplasia.36,41,44 By contrast, other experimental infection studies have not shown an association between infection and gastric pathology.43 A recent investigation of 30 dogs with lymphoplasmacytic gastritis identified Helicobacter in 77%, but there was no correlation between infection and the severity of inflammatory change. The same study quantified cytokine gene expression within these mucosal biopsies and identified a Th1-skewed profile with expression of IL-1β, IL-8, IL-10, transforming growth factor (TGF)-β, and interferon (IFN)-γ, but lack of IL-4 transcription. Expression of IL-10 and IFN-γ was most closely associated with the presence of lymphoplasmacytic infiltration.53 There remains little definitive evidence that Helicobacter have a significant role in the development of canine and feline gastritis.
< div class='tao-gold-member'>
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


