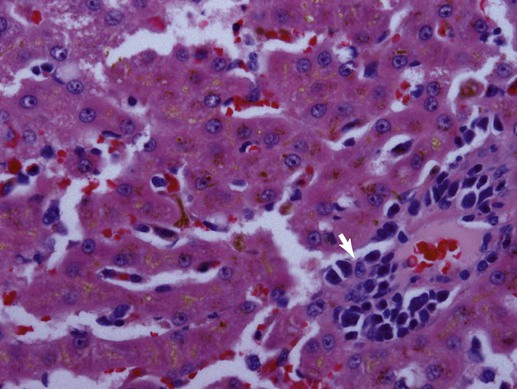
Bacteriologic studies of portal vein blood in mature dogs confirm that alimentary flora can circulate to the liver.32,38,38 Although unproven, it is likely that this also occurs in cats. Bacteria translocating from the gut into the portal vein are eliminated and killed through dual action of hepatic Kupffer cells and activated neutrophils or are simply excreted in bile.18,221 Liver disorders associated with ischemic injury, impaired hepatic artery or cystic artery perfusion, reduced macrophage function, cholestasis, and/or systemic immunosuppression can be complicated by infection with opportunistic enteric flora.25 Hepatoprotection against systemic toxicity and infections can be compromised when the liver is injured in a variety of ways. For example, diffuse liver injury is associated with reduced immune defense, which reflects suppressed bactericidal and opsonic activities, defective chemotaxis, reduced serum complement availability, impaired monocyte function, and abnormal hypersensitivity responses. The integrity of the hepatic reticuloendothelial system (RES, mononuclear phagocyte system) is important in the protection of the liver from enteric bacterial translocation associated with hemorrhagic and endotoxic shock, trauma, bowel ischemia, and surgical manipulations of the intestine or colon. Experimental work and observations in human patients confirms that RES function can be compromised secondary to chronic liver disease, portal hypertension, portosystemic shunting, and cholestasis.163 Consequently, patients with diverse hepatobiliary disorders have increased risk for hepatic infection and polysystemic complications. The liver has additional risk for infection associated with its expansive sinusoidal endothelium that provides a site for invasion by vasculotropic organisms. Substantial host-dependent differences exist in hepatic clearance of bloodborne particulates. In dogs, 60% to 90% of hematogenously borne bacteria and particulates are removed in the liver and spleen, placing each organ at risk for infection or injury.55,246 Conversely, pulmonary macrophages assume these primary functions in the cat. Failure of hepatic Kupffer cell function in the dog can shift the burden of RES surveillance to other organs (e.g., spleen, lungs, and lymph nodes), as shown in animals with chronic liver disease associated with acquired portosystemic shunts and dogs with congenital portosystemic vascular anomalies (PSVA).106,127 Although it is well established that humans with hepatic cirrhosis have increased prevalence of bacterial infections, similar data are unavailable for dogs and cats. Predominant perfusion of hepatic sinusoids with portal venous blood (a low-flow, low-pressure system), rather than arterial blood, facilitates efficient bacterial removal by the RES. The less dynamic flow driven by portal perfusion within the sinusoids, from peripheral lobular portal to centrilobular hepatic veins. This permits greater opportunity for pathogen anchorage to macrophages and their subsequent phagocytosis.114,188 However, portal flow can be compromised as with disorders associated with hepatofugal circulation: chronic liver disease with intrahepatic portal hypertension and acquired portosystemic shunting or PSVA. Increased hepatic arterial perfusion compensatively intensifies sinusoidal blood flow, which potentially thwarts efficient phagocytosis. Endotoxin (lipopolysaccharide [LPS]), a glycolipid derivative from the outer bacterial cell membrane of gram-negative bacteria, is a normal constituent found in portal venous blood where it reflects translocation of enteric bacteria (see Etiology, Chapter 35; Pathogenesis, Chapter 36; and Bacterial Translocation, Chapter 88).215 Normally, hepatic Kupffer cells efficiently clear endotoxin to attenuate systemic exposure. Endotoxin extraction is enabled by LPS-binding glycoproteins on hepatocytes that facilitate transfer to high-affinity LPS receptors on Kupffer cells. Contact with LPS activates Kupffer cells directly via Toll-like receptor signaling or indirectly by triggering complement activation. On exposure to endotoxin, activation of Kupffer cells initiates a multitude of signaling pathways resulting in a spectrum of responses.18 Signaling of proinflammatory cytokines can exacerbate previous liver injury or induce small focal “sentinel” lesions in the hepatic parenchyma. These injuries, such as lipogranulomatous foci, or low-grade inflammatory foci, nonspecifically increase serum liver enzyme activities. Reduced liver function or perfusion can each increase hepatic and systemic exposure to endotoxin. In fact, endotoxemia in the absence of overt sepsis is a common finding in humans with cirrhosis. There is evidence that patients with cirrhosis maintain a markedly imbalanced cytokine response (the so-called cytokine storm involving proinflammatory signals) that converts normally beneficial infection-thwarting responses into excessive, damaging inflammation.100 The highest levels of endotoxemia are associated with hepatic failure, hepatic encephalopathy, and death. However, it is unclear whether systemic endotoxemia reflects normal exposure left unchecked by a dysfunctional liver, relevant systemic abnormalities, or increased enteric endotoxin translocation, or whether endotoxemia plays a causal role in liver disease or merely represents an epiphenomenon. Because enteric bile acids bind and inactivate endotoxin, patients with extrahepatic bile duct occlusion (EHBDO) or severe intrahepatic cholestasis with impaired enterohepatic bile acid circulation have higher risk for endotoxin translocation.206 Clinically healthy dogs, dogs with experimental dimethylnitrosamine-induced chronic liver disease and acquired portosystemic shunts, and dogs with PSVA have measurable endotoxin concentrations in portal and systemic venous blood.108,182 Dogs with induced chronic liver disease associated with intrahepatic portal hypertension and acquired portosystemic shunts develop significantly greater endotoxin concentrations in portal venous, hepatic venous, and caudal vena cava blood, compared to healthy control dogs.108 In the portal vein, median endotoxin concentrations were approximately 17-fold higher in dogs with chronic liver disease.108 In another study, approximately 85% of dogs with chronic liver disease, compared to approximately 40% of healthy control dogs, had detectable endotoxin in venous blood collected from the portal and hepatic veins, and caudal vena cava. Mean endotoxin concentrations in portal vein and systemic venous blood in 10 medically stable, antibacterial treated dogs with PSVA and 5 clinically healthy control dogs were 22.5 ± 8.9 pg/mL versus 17.6 ± 6.0 pg/mL and 28.0 ± 16.9 pg/mL versus 23.6 ± 11.0 pg/mL, respectively.182 Endotoxin concentrations in systemic blood of six dogs with PSVA, with paired pre- and post-ligation (5 to 13 months) blood samples, demonstrated a reduction in endotoxin concentrations after shunt ligation. The mean preligation endotoxin was 19.0 ± 7.0 pg/mL and postligation endotoxin was 9.7 ± 3.7 pg/mL; p = 0.06).182 Small patient numbers may have precluded achieving significant differences. Taken together, observations in clinically relevant conditions suggest that systemic endotoxemia is a real phenomenon in dogs. Also, by inference,increased systemic exposure to enteric microbes and their by-products results from hepatofugal portal venous flow. Systemic response to endotoxemia, the presence of bacteremia, or evidence of altered oxygen utilization in portal blood have not been shown in dogs with PSVA.223 Hepatic Kupffer cells comprise the largest compartment of tissue macrophages in the body, representing 80% to 90% of the total fixed macrophages and approximately 35% of the nonparenchymal liver cells.18,93 Residing mainly within the lumen of hepatic sinusoids and adherent to endothelial cells by long cytoplasmic processes, Kupffer cells are most numerous in the periportal area (zone 1, see later) where they offer first-line defense against bacteria, endotoxin, and microbial debris delivered from the alimentary canal.76 Activation of Kupffer cells by endotoxin and other signaling molecules is mechanistic to protective and disease-producing effects. Kupffer cells possess both Fc and C3 receptors and phagocytize a wide variety of opsonized and nonopsonized particulates.133 Similar to other mononuclear phagocytes, Kupffer cells also have the capacity to function as antigen-presenting cells, for induction of T lymphocytes and, on activation, can release superoxide radicals, hydrogen peroxide, nitric oxide, hydrolytic enzymes, and eicosanoids (prostaglandins and leukotrienes) that can assist in antigen destruction. They also release a large number immunoregulatory and inflammatory cytokines, including interleukin (IL)-1, IL-6, tumor necrosis factor (TNF)-α, platelet-activating factor, transforming growth factor-β, and interferon-γ. TNF-α is a major early mediator in the systemic inflammatory response syndrome associated with gram-negative sepsis.130,235 This mediator is partially responsible for a number of pathophysiologic hepatic responses, including the acute-phase response, hyperlipidemia, free oxygen radical formation, fibrinogenesis, and intrahepatic or molecular cholestasis of sepsis.29,73,130,192 The transcriptional regulatory factor, nuclear factor kappa-B (NF-κB), plays a central role modulating immunoregulatory mediators involved with oxidative stress and sepsis.237 In fact, activity of NF-κB is generally considered a central mediator of stress responses. In liver, NF-κB is induced in response to ischemia-reperfusion injury, hemorrhagic shock, liver regeneration, and certain chemotherapeutic agents. A heterogenous population of Kupffer cells resides in different lobular zones (e.g., zone 1 in periportal, zone 3 in periacinar, adjacent to hepatic vein, and zone 2 in hepatic parenchyma spanning between zones 1 and 3). Kupffer cells associated with zone 1 are larger, more phagocytic, and generate greater amounts of TNF-α, IL-1, prostaglandin E, and lysosomal enzymes. Smaller Kupffer cells are associated with zone 3, where more nitric oxide and superoxide are produced; these exhibit greater cytotoxic activity to some stimuli and are more easily activated.69 Nitric oxide released by Kupffer cells mediates a wide variety of physiologic events, including vasodilation, neutrophil chemotaxis, and adhesion of neutrophils to vascular endothelium (response to bacteria or endotoxin).93 As potent inducers of inflammatory cytokines, Kupffer cells are implicated in pathologic events resulting in liver injury.215 Inflammatory mediator release from Kupffer cells is enhanced, after exposure to endotoxin or to iron, in reactive hepatobiliary processes. Later, during the course of an infection, other cells including hepatocytes, T lymphocytes, and immigrating phagocytes (monocytes and neutrophils) contribute cytokines to the inflammatory process. Some bacteria undergo initial physical attachment to Kupffer cell surface receptors (macrophage scavenger receptors) and are subsequently cleared from the sinusoidal circulation by microbicidal neutrophils and macrophages. These receptors have high affinity for a broad range of polyanionic ligands including lipoteichoic acid, a cell wall component of gram-positive bacteria.69,177 However, other organisms are not efficiently cleared by Kupffer cells (e.g., Pseudomonas aeruginosa, Morganella morganii, and Serratia marcescens) because of unique cell surface components or increased hydrophobic tendencies, or both.92 In summary, Kupffer cells contribute to cellular defense from microbial invaders through collaborative interaction with neutrophils or by independent activation of the complement pathway. Depending on clearance mechanisms, intercellular signaling, and the host response, Kupffer cells also induce inflammatory mediators that perpetuate chronic hepatitis. Routine, uneventful elimination of bacteria, endotoxin, and particulate or antigenic debris occurs by collaboration between Kupffer cells and infiltrating neutrophils.92 As described previously, acute bacterial clearance involves extracellular binding of microbial carbohydrate residues to Kupffer cell lectins. Complementary adhesion molecules expressed on neutrophils anchor them to Kupffer cells, facilitating phagocytosis (internalization, killing) of adhered bacteria. Neutrophil participation in the clearance process also can impart self-induced injury. In this regard, a distinguishing feature of endotoxemia and sepsis is neutrophil accumulation in hepatic sinusoids where they can foster production, release, and accumulation of toxic metabolites, free radicals, and degradative enzymes (reactive oxygen intermediates and proteolytic enzymes). Neutrophil involvement can cause histologic misinterpretation of the host response, by sometimes erroneously suggesting involvement of infectious organisms. Cell injury ascribed to focal accumulations of neutrophils contribute to lesions characterized as reactive hepatitis, commonly observed in liver biopsy specimens collected from animals with inflammatory bowel disease (IBD) and other systemic inflammatory conditions. Such foci may represent sequelae of a normal hepatic sentinel response. Neutrophil-associated tissue injury is normally curtailed by cell apoptosis and phagocytosis by Kupffer cells after infection control or toxin elimination. Dysregulation of these control mechanisms can contribute to continued chronic liver injury and inflammation. Cholangiocytes in larger bile ducts express the polymeric IgA receptor (secretory component) that localizes secretory IgA (S-IgA) to the ductal system. Uptake from the basolateral side of the cholangiocyte is followed by transport to the luminal ductal surface, where secretory component is cleaved from but secreted with S-IgA into bile. Cholangiocytes from larger intrahepatic bile ducts, septal bile ducts, interlobular bile ducts, and bile ductules express Toll-like receptor 2, 3, 4, and 5 that bind a variety of ligands including bacterial molecules, double-stranded messenger RNA (mRNA), LPS, and flagellin.101 In the presence of inflammatory cytokines, cholangiocytes also can activate the (proinflammatory) NF-κB pathway. Cholangiocytes also produce the antimicrobial peptides defensin and cathelicidin, part of the innate immune system, that defend the biliary tree against gram-positive and gram-negative bacteria, mycobacteria, fungi, and viruses. Expression of cathelicidin in biliary epithelium is enhanced by exposure to endogenous and therapeutic bile salts (e.g., ursodeoxycholic acid). In biliary inflammation, cholangiocytes also recruit CD4+ T cells as well as immature dendritic cells to provide defense against antigenic and microbial challenge. Cholangiocytes also express a variety of leukocyte and vascular adhesion molecules that can facilitate tissue-specific leukocyte migration, slowing leukocyte circulation near damaged epithelium and trafficking cells to the targeted site of injury. Expression of antigen-presenting molecules on cholangiocytes also may contribute to local immune defense. Hepatobiliary production of bile, S-IgA (two IgA molecules and a peptide J chain), and the secretory component importantly contribute to the health and integrity of the biliary and gastrointestinal (GI) systems. S-IgA is the major immunoglobulin in bile; IgG and IgM are present in much smaller quantities. Bile contains approximately twice the concentration of S-IgA as found in upper intestinal fluid.50 During transit, secretory component prevents proteolytic digestion of IgA in the alimentary canal with its carbohydrate residues anchoring S-IgA to mucus. There, S-IgA binds and neutralizes bacterial toxins and prevents bacterial adhesion to the mucosal interface. S-IgA also neutralizes intracellular microbes and microbial products during enteric translocation. Immune complexes of S-IgA and foreign antigen(s) (e.g., bacteria, viruses) in the lamina propria of the gut also can undergo secretory component-facilitated capture-transport back into the alimentary lumen. There, potential pathogens are delivered to the hostile proteolytic mucosal environment, thereby preventing their cell invasion, infection, and translocation. Bile and IgA each influence the enteric bacterial population (type, number of organisms, and enterocyte adherence). Normal physiologic choleresis mechanically cleanses the biliary pathways of microbial organisms and particulate debris. This function is thwarted in cholestatic disorders. A normal biliary-enterobacterial cycle permits rapid elimination of bacteria that have entered the biliary system where locally produced IgA prevents epithelial invasion.30,205 The composition of bile salts contributes a synergistic influence with IgA binding, limiting enteric and biliary bacterial translocation. In health, tight junctions between hepatocytes (abutting canaliculi) resist bacterial entry into canalicular bile. High competence of the extrahepatic biliary structures against microbial penetrance and the normal pressure differential maintained in the biliary system limit retrograde access of enteric organisms into the liver. However, development of EHBDO or other processes fostering cholestasis can disrupt normal protective mechanisms, increasing host susceptibility to opportunistic infection.205 The importance of enteric bile and S-IgA in protecting against bacterial translocation is supported by in vivo studies using experimental rodent models. The prevalence of bacterial translocation (mesenteric lymph nodes with positive culture results) increases from 33% in animals subjected only to visceral surgical manipulation to 48% in EHBDO and to 65% in animals with biliary decompression by external drainage.209 The microbial composition of the portal venous circulation, hepatic tissue, and bile in healthy patients has been investigated for more than 60 years. Older studies suggest that liver tissue is commonly contaminated, especially with Clostridium spp.; however, other work is contradictive.53,66,66 In the absence of biliary tree obstruction or choleliths, the gallbladder and bile are usually sterile. Improved methods of collecting samples to reduce cross-contamination likely explain disparate observations between older and more recent investigations. However, if biopsy specimens or bile from clinically healthy animals is inoculated into enrichment media that facilitates growth of more fastidious organisms, positive culture results are more common, and discerning relevant from irrelevant findings becomes more difficult.165 Normally, enteric organisms delivered to the liver that are not extracted and killed by cooperative neutrophil–Kupffer cell interactions are excreted in bile. Even in clinically healthy animals, portal venous translocation of large bacterial inocula result in hepatic sinusoidal bacteremia and bacterobilia.215 In the diseased liver (e.g., perfusion abnormalities, impaired macrophage function, cholestasis), survival of infectious agents may be permissively allowed by loss of normal protective mechanisms and by the presence of cholestasis.30 Consequently, diseases of the biliary tract and liver may be complicated by colonization with bacteria as a secondary phenomenon. Positive bacterial culture results from liver tissue or bile from animals with chronic liver disease, not thought to be initiated by bacterial infection, support this contention. Because the liver plays a key role in protecting the systemic circulation from gram-negative sepsis, a variety of liver disorders increase host risk for infection with opportunistic enteric pathogens. The enhanced pathogenicity of gram-negative sepsis has been confirmed in experimental models of cirrhosis.105 Bacterial organisms isolated from bile, gallbladder or liver in humans and animals with hepatobiliary disease most commonly represent enteric flora. Box 89-1 summarizes the prevalence and type of organisms isolated from the affected sites of dogs and cats with hepatobiliary diseases.9,139,185,245 Notably, a similar spectrum of bacterial isolates exists between dogs, cats, and humans .239,245 The greatest risk for infection and postoperative sepsis is associated with cholestatic disorders (e.g., EHBDO, cholecystitis, cholangitis), chronic liver disease associated with portal hypertension, disorders compromising hepatic perfusion or Kupffer cell function, and conditions promoting enteric bacterial translocation (e.g., IBD, bowel obstruction, enteric foreign body, bowel ischemia, endotoxic shock). Box 89-2 lists the mechanisms contributing to or amplifying infections.* High concentrations of plasma, tissue, and bile dihydroxy bile acids associated with cholestasis alter lymphocyte-mediated immune responses.206 Furthermore, studies confirm that hepatic endotoxemia suppresses natural killer cell responses in the liver, secondary to induced interferon-γ. This increases susceptibility to splanchnic-acquired bacteremia as natural killer cell function is an integral component of innate immune protection.118 Any disorder invoking cholestasis can compromise protective mechanisms derived from bile and physiologic choleresis. Along with these, EHBDO provokes additional changes that facilitate hepatobiliary and systemic bacterial infection. Animal models of EHBDO convincingly demonstrate that impaired enteric bile flow favors small intestinal bacterial overgrowth (SIBO) and enteric bacterial translocation to mesenteric lymph nodes, liver, and the systemic circulation.48 Cessation of enteric bile delivery (bile salts, S-IgA) eliminates the suppressive influence of bile salts on endogenous bacterial flora and S-IgA on bacterial mucosal adherence. Defective RES function and altered sinusoidal fenestra near the peribiliary plexus (within the portal triad) increase bacterial access to the hepatic circulation and biliary tree. Numerous factors heighten the risk for hepatobiliary infection and development of bacterial cholangitis by any route: ascending, hematogenous, or lymphatic.174,206 Although surgical, percutaneous, or stent methods of achieving biliary decompression can ameliorate jaundice short-term in EHBDO, chronic injury to biliary structures, associated with functional changes, may only reverse slowly, with some functions never being fully restored. Immune dysfunction in cholestatic liver injury permissively augments infections. These infections are a result of inadequate or inappropriate antigen processing by the RES, overproduction of cytokines by Kupffer cells, abnormal Kupffer-hepatocyte interactions, and the altered resident population of natural killer cells. Decreased Kupffer cell activation to endotoxin increases risk for endotoxemia during episodes of heightened exposure (e.g., enteric hemorrhagic gastroenteritis, surgical manipulations of viscera). Hepatofugal portal circulation, associated with acquired portosystemic shunts or intrahepatic shunting through collagenized sinusoids, reduces hepatocellular exposure and processing of portal-borne substances, thereby increasing systemic exposure to immune complexes, enteric bacteria, and antigens normally sequestered within the liver. Impaired hepatic bacterial opsonization thwarts effective macrophage bacterial clearance and heightens risk for infection. Dysregulation of inflammatory cascades, crucial to wound healing, may increase postoperative complications, including wound dehiscence and infections that compromise surgical recovery.206 Experimental models have helped clarify the physiologic significance of enteric portal bacteremia. Infusion of 105 to 107 bacteria into the splenic vein of normal cats significantly retards bile flow and causes bacterobilia within 30 minutes. However, in the presence of EHBDO, only modest bacterial inocula (103) lead to bacterobilia within the same time frame. As bacteria enter sinusoidal blood, some are phagocytized (Kupffer cells and neutrophils) and killed while others remain viable entering bile.221 Experimental studies confirm that any cause of cholestasis can augment development of bacterobilia, including the cholestasis resulting from endotoxemia. That portal bacteremia is an important source of bacterobilia is reflected in the distribution of organisms most often isolated from hepatic and biliary cultures (see Box 89-1).239,245 Transient or episodic portal bacteremia likely explains the unexpected isolation of bacterial organisms from liver or bile in animals with illnesses thought not to be primarily bacterial in origin (Box 89-3), especially if samples are collected after surgical manipulations of visceral structures. The idea that translocation of bacteria and endotoxin from the GI tract initiate or exacerbate systemic infections has been increasingly accepted as the so-called gut hypothesis of sepsis and multiple organ failure.60 Increased vulnerability of a jaundiced host to gut-barrier breakdown and bacterial translocation is confirmed by many experimental studies and is strongly supported by clinical observations in veterinary patients. Diseases involving the biliary tract especially thwart protective mechanisms preventing bacterobilia.217 Studies in dogs undergoing routine ovariohysterectomy (OVH) prove that enteric bacterial translocation occurs even in healthy individuals.53 In one study, bacterial translocation was implicated by positive culture results of a single mesenteric lymph node from each of 52% of 26 dogs. The number of bacteria cultured varied from 50 to 105 organisms per gram of tissue. In decreasing order of prevalence, isolated organisms included Escherichia coli (n = 6), Bacillus (n = 5), nonhemolytic Streptococcus (n = 4), Salmonella (n = 3), coagulase-negative Staphylococcus (n = 2), Enterococcus (n = 2), and one each with Staphylococcus pseudintermedius, Clostridium sordellii, Micrococcus spp., Pseudomonas spp., Lactobacillus spp., and Propionibacterium acnes. There were no bacteria isolated from portal blood samples. Another study involved 20 dogs: 13 healthy females scheduled for routine OVH, and 7 healthy males presented for resolution of umbilical hernias (n = 4), or removal of gastric foreign bodies (n = 2) or mesenteric lipoma (n = 1).165 Of these dogs, one-half had histologic liver lesions. Needle biopsy samples (14-guage diameter) were inoculated onto blood agar and into enrichment broth (aerobic) and Wilkins-Chalgren anaerobe agar and anaerobic enrichment broth. A minimum of five colonies designated a positive culture result. Of 10 dogs lacking liver lesions, 50% had positive liver culture results with one organism, including Staphylococcus xylosus, E. coli, Pseudomonas cepacia, Staphylococcus auricularis, Staphylococcus epidermidis, Clostridium subterminale, and Clostridium perfringens. Reconciliation of culture results from clinically healthy dogs compared to those with hepatic disease indicates that adjunctive review of culture results, clinical signs, cytologic impression smears that can reveal suppurative inflammation and bacteria, and histopathologic features of biopsied tissue is necessary to determine the relevance of a positive aerobic or anaerobic bacterial culture result. Translocated enteric bacteria and endotoxin activate Kupffer cells, initiating a cascade of effects that can lead to hepatic fibrosis, such as cytokine secretion, chemotaxis, vascular adhesion, and degranulation of neutrophils, and proinflammatory changes involving sinusoidal endothelium and hepatic stellate cells. The latter cells are the source of connective tissue that produces fibrosis in chronic liver disease. Tissue damage reflects injury derived from reactive oxygen species, cytokines, and proteases, along with reactions involving complement and activated coagulative proteases and platelets. Correlation of plasma endotoxin concentrations with morbidity and immunosuppression is variable and inconsistent as a result of differences in regional blood sample collection, methods of endotoxin detection, lack of endotoxin standards, and expression units.89,186 Enteric translocation of bacteria and subsequent hepatobiliary invasion is enhanced in the presence of (1) bowel disease (direct mucosal injury), (2) altered gut flora with gram-negative microbial overgrowth (SIBO), (3) portal hypertension, (4) splanchnic hypoperfusion, (5) hepatofugal portal circulation (acquired and congenital portosystemic shunting), (6) local or systemic immunosuppression, including impaired macrophage function, (7) altered gut motility (slow transit time documented in cirrhosis), (8) absence of enteric bile (bile acids, S-IgA, mechanical cleansing function of bile in the biliary tree), and (9) surgical visceral manipulations. For example, humans and experimental animals with chronic liver disease demonstrate a reduced enteric transit rate that increases risk for SIBO and enteric bacterial translocation.181 Contributing factors include portal hypertension, acquired varices, gastroduodenal vascular ectasia (portal hypertensive gastroenteropathy), and oxidative bowel damage that may or may not be associated with IBD.44,202,202 Although increased propensity for enteric bacterial translocation in EHBDO is proven in dogs and cats, the utility of preoperative biliary decompression in improving patient survival remains controversial.44,221 The clinical impact of cholestasis on enteric bacterial and endotoxin translocation is not clearly exemplified by response in humans to preoperative biliary tree decompression, where endoscopically facilitated interventions are routine. Several controlled retrospective studies and meta-analyses confirm higher overall septic complications in patients subjected to preoperative decompression as compared to patients directly proceeding to surgery.207,234,234 Increased risk for infection is associated with procedural trauma and insertion of plastic stents that are compromised by development of biofilm (bacteria, organic debris, calcium salt crystallization) and obstructive debris that induce septic cholangitis.. Bacterobilia may be clinically silent until biliary obstruction leads to systemic sepsis by biliary-venous reflux. Increased pressure (at or above 25 cm water) in the biliary system causes retrograde flow of bile (regurgitation) into hepatic sinusoids and is a proven prerequisite for retrograde infection. The importance of mechanical disruption of bile flow to biliary tree infection is well exemplified by long-term follow-up of humans with choledochoduodenostomy where retrograde bacterial invasion of the biliary tree is the rule. In the presence of a stoma stricture (postoperative cholenterostomy scarring) or stagnant loop (sump syndrome in which a pocket develops at the anastomotic site), retrograde bacterial infection, cholangitis, and cyclic illness follow. A small study in dogs with biliary enteric anastomosis confirmed persistent infection in dogs with cholecystoduodenostomy and cholecystojejunostomy anastomoses within 1 to 6 months of biliary diversion.150 However, these human patients generally do not develop illness associated with septic cholangitis as long as mechanical obstruction of bile flow is averted. Clinical and experimental evidence suggests that symptomatic biliary infections are most likely when obstruction is incomplete or intermittent and are potentiated by the presence of a foreign body such as a cholelith or polypropylene (plastic or rubber) biliary stent.65,219 Experimental studies in cats confirm that portal venous bacteremia, induced by infusion of large numbers of pathogenic bacteria, can cause bacterobilia and that cats with EHBDO are more vulnerable.221 The presence of bacteria within choleliths also can sustain bacterial infections in the biliary tree.104 Mechanical resolution of EHBDO mobilizes sequestered bacteria, resulting in sudden appearance of bacteria in bile and potentially in the systemic circulation. Surgery-associated trauma also increases bacterial translocation, more so in EHBDO, compared to patients lacking cholestasis. Thus, jaundiced patients with cholestasis undergoing major abdominal surgery may be compromised not only by the stress of surgery but also by perioperative release of endotoxins and bacteremia. Because endotoxin can induce bacterial translocation by itself, this may sustain the cycle of splanchnic bacteremia.61,64,64 Failure to provide adequate antibacterial coverage during perioperative and postoperative periods can significantly increase risk of postsurgical infection and sepsis in patients with biliary tree infections and EHBDO. Importantly, surgical trauma by itself augments risks imposed by reduced gut-barrier function in cholestasis and/or hepatobiliary dysfunction.62,178 It has been argued that presurgical internal drainage of the biliary tree in EHBDO reduces postoperative septic complications.144 However, as described previously, it is difficult to reduce these risks because of trauma associated with stent insertion and complications associated with stent function. Modifying the enteric microbial population with antibacterials (e.g., quinolones, neomycin, tobramycin, rifaximin) or using certain probiotics (lactobacilli combined with antioxidants) has reduced septic complications in humans and experimental animals with cholestatic liver disorders. In one study, treatment of cirrhotic humans with ciprofloxacin reduced risk for spontaneous bacterial infection with enteric organisms.190 Once the biliary-enteric communication has been surgically or endoscopically opened, many of the bacterial protective mechanisms lost from cholestasis are abruptly reestablished.206 Despite the immense potential for hepatic exposure to infectious organisms, increased liver enzyme activities and hepatic dysfunction in infectious disease most often reflect secondary effects of systemic infection on the liver, rather than specific hepatic involvement.158,167 Pyrexia, anoxia, nutritional deficits, released toxins, and inflammatory mediators each contribute to clinicopathologic abnormalities. “Innocent bystander” injury from pathologic conditions initiated elsewhere in the body can lead to inappropriate diagnostic focus on the hepatobiliary system. Occasionally, self-perpetuating chronic hepatitis follows as a complication of infection with bacterial or viral agents. Examples include chronic hepatitis in dogs after infection with Leptospira or canine adenovirus-1 or after the inadvertent parenteral injection of intranasal Bordetella bronchiseptica vaccine (see Chapters 42, 4 and 6, and 100).20,85,85 An emerging role of Helicobacter spp. in humans with cholestatic liver disease, cholecystitis, and neoplasia of the biliary tree suggests that a relationship may also exist between these organisms and liver disease.77,136,136 Isolation of Helicobacter canis from a single dog with multifocal necrotizing hepatitis has been reported; organisms were observed using a silver stain at the periphery of necrotizing lesions.78 Bacteria were concentrated between adjacent hepatocytes in bile canaliculi and observed in the lumen of bile ducts. Organisms were cultured and phenotypically and molecularly identified as being different from H. canis. However, detection of Helicobacter DNA using polymerase chain reaction (PCR) and amplicon sequencing from archived formalin-fixed liver tissue was possible in only 2 of 32 cats with cholangiohepatitis (1 with mild lymphocytic, 1 with suppurative cholangiohepatitis) and in 1 of 17 control cats with noninflammatory liver disease (PSVA).95 Based on comparisons to published sequence homologies, Helicobacter pylori in combination with Helicobacter nemestrinae, or H. pylori, in combination with Helicobacter felis-Helicobacter cinaedi, were detected in two cats with cholangiohepatitis. Helicobacter bilis was identified in the cat with PSVA. In no case were organisms visually identified in tissue by silver stain or immunocytochemistry. Another survey from cats with lymphocytic cholangitis detected Helicobacter species in bile by PCR in 4 of 15 (27%) cats with cholangiohepatitis, compared to 7 of 12 (58%) clinically healthy control cats.23 These results suggest either enteric contamination of biliary samples or enterohepatic-biliary circulation of bacterial organisms or remnant DNA. For a further discussion of Helicobacter infections, see Chapter 37. Association of hepatic dysfunction and cholestatic liver injury is documented in humans and in numerous animal models as a consequence of systemic bacterial infection and endotoxemia (see Chapter 36).66 In addition to endotoxin-induced hepatic injury, patients with obstructive jaundice, cirrhosis, or following extensive hepatic mass excision have heightened susceptibility to endotoxemia due to reduced Kupffer cell function, impaired hepatic perfusion, and enhanced enteric microbial translocation.130 Thus, determining the precise cause or effect relationship between endotoxemia, sepsis, and liver disease may be difficult. Formation of bile is mediated and maintained by coordinated function of a set of membrane transporters located within hepatocytes, cholangiocytes, and enterocytes.130 Bile acids and glutathione (GSH) are the main solutes driving bile formation, and impairment of these bile acid–dependent and bile acid–independent mechanisms causes cholestasis. Function of the pump systems responsible for bile formation is regulated at multiple levels, from initiation of RNA transcription in the nucleus to microdomain localization in cell membranes.130 Multiple steps are necessary for proper function and localization of the pathway-transporter proteins operative in bile formation. These steps are involved with intracellular signaling and transport pathways, cytoskeletal structures, and signaling regulators that modify protein activity after transcription, as well as localization and stability.130 Because this system also conducts biotransformation reactions involving potentially damaging and bioactive small molecules, downregulation puts hepatocytes at risk for secondary damage from reactive intermediates and toxins. Expression and function of key molecular components are involved in adjusting bile flow as a response to cell injury or various “stressors.” Such events lead to downregulation of hepatobiliary transport pathways culminating in cholestasis. Such is the case with gram-negative sepsis and endotoxemia where inflammation drives a cholestatic sequela. Cholestasis also can be induced secondary to infection with gram-positive and other microbial organisms. Endotoxin downregulates hepatic transport systems involved with bile acid uptake and excretion as well as phase I and II detoxification systems. This impairs bile formation and leads to accumulation of bile acids and toxins in both liver and the systemic circulation.* Major pathways involved are considered acute-phase reactants and include TNF-α, IL-1β, and IL-6 released from LPS-activated Kupffer cells, sinusoidal endothelial cells, hepatocytes, and cholangiocytes. Sepsis also induces release of additional small molecules (e.g., nitric oxide) that lead to local oxidative injury, hypotension, vascular collapse, and cholestasis. Endotoxin has been shown to influence hepatic gene expression through numerous intersecting signaling pathways with major effects mediated through Kupffer cells by activation of NF-κB and adapter protein-1. These proteins secondarily induce the acute-phase response influence, described above. Inflammatory signals directly initiated by LPS or indirectly via effector cytokines reduce expression of hepatocellular transporters at both the gene and protein level influencing the principal bile acid transporter and bile salt export pump located in the basolateral and canalicular membranes, respectively.130 There is sufficient evidence linking endotoxin and endotoxin-induced cytokines to cholestasis.10,130,130 In sepsis and clinically evident endotoxemia, hyperbilirubinemia is a central clinical finding often disproportionate to the magnitude of serum aminotransferase and alkaline phosphatase (ALP) activities. Serum bile acid concentrations also are increased in sepsis and endotoxemia reflecting transporter downregulation. Intrahepatic cholestasis induced by severe extrahepatic bacterial infection, experimentally induced in dogs and cats, has been clinically recognized (Fig. 89-1).221,225 Hepatic response to systemic infection has been studied in dogs experimentally infused with endotoxin or live gram-negative bacteria, or both.74,96,98,99 Acute morphologic changes include dilation and congestion of sinusoids and hepatic veins, central (zone 3) and midzonal (zone 2) hepatocellular necrosis, fatty or vacuolar degeneration (not glycogen associated), acute diffuse influx of inflammatory cells (neutrophils and monocytes), and microabscess formation. Kupffer cell hyperplasia is occasionally described, and canalicular stasis (microscopically evident “bile plugs”) observed with chronicity. Considerable hepatocellular dysfunction can cause a shift to anaerobic metabolism, impairing gluconeogenesis and mobilizing triglycerides from adipose stores. In dogs, acutely increased serum triglyceride, nonesterified fatty acids, and cholesterol concentrations reflect the metabolic shift to fatty acid oxidation.96 If this activity also occurs in cats, peripheral fat mobilization might augment development of hepatic lipidosis. In fatty liver modeled in the choline-deficient rats, impaired RES function increases host susceptibility to endotoxemia.166 Although it is not known if this is also true in cats with hepatic lipidosis, the common occurrence of a more primary disease causing anorexia and subsequently hepatic lipidosis warrants consideration. For example, endotoxemia derived from underlying IBD or constipation might complicate treatment of some cats with hepatic lipidosis. Animals with compromised liver function or cholestasis experiencing gastroenteric hemorrhage have increased risk for endotoxemia. Especially relevant is greater risk for endotoxemia in patients with portal hypertension complicated by ascites, acquired portosystemic shunts, and portal hypertensive encephalopathy. In these, enteric health can be significantly affected by an associated hypertensive vasculopathy. This acquired mucosal perfusion defect is characterized by noninflammatory mucosal and submucosal vascular dilation, ectasia, and edema; bowel wall erythema and friability; and a propensity toward gastroduodenal ulceration. In some cases, multifocal enteric bleeding leads to critical blood loss and hepatic encephalopathy (secondary to enteric hemorrhage and hemoglobin digestion). SIBO is reported in approximately 60% of humans with cirrhosis when reduced small intestinal transit rate and impaired mucosal defenses are contributing factors.15 Additionally, certain commensal bacteria are known to modify small intestinal and colonic permeability in human and animal cirrhosis; the greatest permeability change occurs if there is associated portal hypertension. In cirrhosis, collective changes contribute to heightened risk for endotoxemia when enhanced enteric permeability and endotoxemia are implicated as initiators of cytokine and inflammatory mediator production in liver, gut, mesenteric lymph nodes, and systemic circulation. Accompanying enhanced gut bacterial and endotoxin translocation, impaired clearance secondary to portosystemic shunt formation and reduced RES phagocytic activity in the body foster development of systemic infection and sepsis. Enteric malabsorption and loss of albumin and other transport and anticoagulant proteins (e.g., antithrombin, protein C, α1-antitrypsin) compromise the defense against infection, body fluid partitioning, and the ability to maintain an anabolic condition essential to thwart development of hepatic encephalopathy. These patients should be treated with broad-spectrum antimicrobials appropriate for enteric opportunists. However, it should be considered that certain antimicrobials actually augment endotoxin release (Box 89-4).138 Paradoxic endotoxin release and related clinical signs are explained by a Jarisch-Herxheimer-like reaction that temporarily worsens the clinical status of the patient undergoing therapy for infections.56,110,110 Risk relates to the antibacterial mechanism of action and whether the microbial cell wall remains intact on antibacterial exposure. During active infection, endotoxin is spontaneously released during multiplication of microorganisms and secondary to bactericidal immune responses. However, endotoxin release can be intensified by antibacterial-induced disintegration of bacterial cell walls, a biologic effect that can generate a 50-fold greater endotoxin exposure than simple infection. Drugs capable of enacting this response inhibit cell wall synthesis and are bactericidal; these enhance the release of greater amounts of endotoxin relative to antimicrobials that merely inhibit bacterial protein synthesis, such as aminoglycosides. For β-lactam antibacterials, endotoxin-enhancing properties relate to the affinity of the penicillin-binding proteins (PBPs) in the bacterial cell wall. There are three important high-molecular-weight PBPs, numbered according to molecular weight. However, interference with any single PBP does not necessarily induce microbial death; rapid bacterial killing is the optimal therapeutic target and is achieved by inhibition of all three PBPs simultaneously. Unfortunately, many β-lactam antibacterials induce formation of aberrant, viable bacterial forms, depending on antibacterial concentration, antibacterial molecular configuration, PBP-binding specificity, and interaction with bacterial PBPs. Interaction with PBP-1 at concentrations greater than the minimum inhibitory concentration causes rapid and extensive bacterial death (e.g., cephaloridine, cefsulodin), whereas antibacterials with high PBP-3 affinity lead to filament forms of bacteria, with incomplete bactericidal activity (i.e., incomplete lysis). When these drugs primarily interact with PBP-3, they induce formation of organisms with high endotoxin biomass that appear as polymorphic filamentous forms. Examples include piperacillin, mezlocillin, and low concentrations of cefuroxime, ceftazidime, and cefotaxime. Eventual lysis of high-biomass filamentous bacterial forms leads to high endotoxin release. β-Lactams targeting PBP-2 cause formation of round cells called spheroplasts. Spheroid formation is followed by bacterial lysis without extensive endotoxin release; imipenem and meropenem show greatest PBP-2 affinity. These drugs have intermediate potential for endotoxin release during treatment. Because carbapenems (e.g., imipenem, meropenem) are potent inducers of chromosomal β-lactamases, these can induce resistance to other β-lactam antibacterials and should not be used for initial empiric therapy. Other drugs have low potential for endotoxin release. Aminoglycosides induce relatively low levels of endotoxin and have a neutralizing effect. Quinolones (e.g., ciprofloxacin) have a lower endotoxin-liberating potency compared to imipenem or ceftazidime. Glycopeptides such as vancomycin appear to downregulate the biologic effect of endotoxin. Clinical studies in human patients suggest that the degree of endotoxin release initiated by antimicrobial therapy has relevance to clinical recovery, patients with marked prodromal endotoxemia having a poorer prognosis. A summary of antibacterial drugs, and their relative potential to facilitate endotoxin release, is listed in Box 89-4.
Hepatobiliary Infections
Antibacterial Defense
Susceptibility to Infection
Endotoxin
Role of Kupffer Cells
Role of Neutrophils
Role of Cholangiocytes
Bile: Protection from Infection
Bacterial Flora of the Hepatobiliary System
Factors Increasing Risk for Hepatobiliary Infection
Influence of Cholestasis on Hepatobiliary Infections
Transmural Passage of Enteric Organisms
Bacterobilia
Risks Increasing Postoperative Sepsis
Innocent Bystander Effects on the Hepatobiliary System
Systemic Infections
Etiology
Sepsis and Endotoxemia
Hepatobiliary Infections