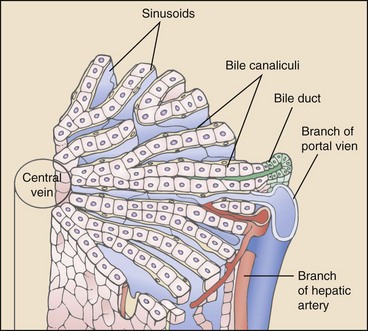
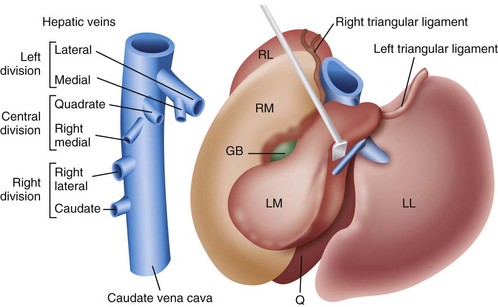
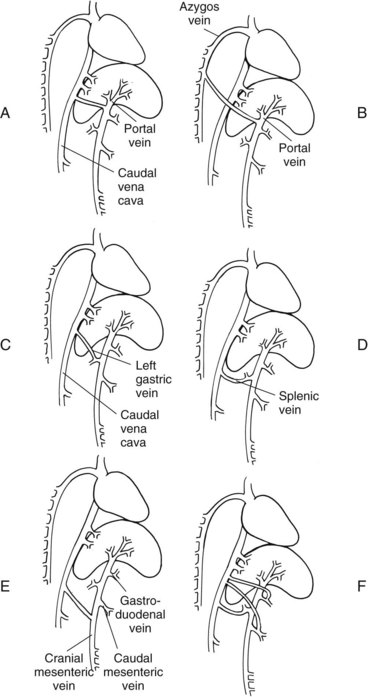
Chapter 96 Abnormalities in portal circulation result in inadequate hepatic development; decreased protein metabolism and production; reduced endogenous and exogenous toxin clearance and drug metabolism; reticuloendothelial dysfunction; altered fat metabolism; and, if severe, progressive liver failure.* As a result, clinical signs such as hepatic encephalopathy, chronic gastrointestinal signs, lower urinary tract signs, recurrent infection, coagulopathies, and delayed growth or poor body condition may occur. This chapter discusses congenital and acquired portosystemic shunts (PSS), hepatic arteriovenous malformations, and microscopic hypoplasia of the portal system. The portal vein supplies 75% to 80% of the afferent blood volume and 50% of the oxygen to the liver, with the remainder supplied by the hepatic artery.91 Tributaries of the portal vein, from caudal to cranial, include the mesenteric vessels, which drain the small intestines and form the cranial mesenteric vein; caudal mesenteric vein, which drains the colon and proximal rectum; splenic vein, which receives blood from the spleen and from the stomach via the left gastric vein; and, in dogs, gastroduodenal vein, which drains portions of the pancreas, duodenum, and stomach (Figure 96-1).52,182 The cranial mesenteric vein is the largest portal vein tributary. In dogs, the portal vein divides into right and left portal branches 0.5 to 1.0 cm beyond the entrance of its gastroduodenal tributary (Figure 96-2). The right portal vein is a short venous trunk that supplies the caudate process of the caudate lobe and the right medial liver lobe; at its bifurcation, it may be partially or completely surrounded by hepatic tissue.182,186 The left portal vein is larger and longer than the right portal vein. It gives off a central branch to the right medial lobe and a small papillary branch to the papillary process of the caudate lobe before dividing into quadrate, left medial, and left lateral branches.182,186 In 60% of dogs, the left portal vein also provides minor blood supply to the dorsal part of the right lateral liver lobe near the porta hepatis.186 In cats, the portal vein divides into right, central, and left branches. In dogs, the hepatic artery divides into right lateral, right medial, and left branches. The presence of additional branches varies.186 The right lateral branch of the hepatic artery supplies the caudate and right lateral hepatic lobes.51 The left hepatic artery branch is usually found on the dorsal or caudal aspect of the left portal branch. It gives rise to the cystic artery and branches to the left lateral and quadrate lobes and part of the left medial lobe.186 The right medial branch supplies the right medial lobe, dorsal part of the quadrate lobe, and a portion of the left medial lobe. In 45% of dogs, the right medial branch arises directly from the hepatic artery and supplies the right medial and quadrate lobes and the gallbladder. In another 50% of dogs, the right medial branch arises from the left hepatic artery and supplies only the right medial lobe.186 Bile ducts and branches of the hepatic artery are usually located on the ventral surface of the portal vein, although some arterial branches may be found dorsally.182 Blood from the portal vein and hepatic artery branches mixes within the hepatic sinusoids before collecting in central veins (Figure 96-3). These central veins merge and eventually form hepatic veins that drain into the abdominal portion of the caudal vena cava.91 Dogs usually have five to eight hepatic veins that form a partial spiral around the caudal vena cava (Figure 96-4). Descriptions of the anatomy of these veins vary among reports.* The left hepatic vein, which is the largest and most cranial of the hepatic veins, terminates on the left lateral aspect of the caudal vena cava near the visceral surface of the diaphragm. About one third to one half of the left hepatic vein’s circumference is encompassed by hepatic parenchyma or obscured by the left triangular ligament. It is unclear from the literature whether the left hepatic vein is a confluence of vessels that drain the left lateral, left medial, quadrate, and right medial lobes or simply the left lateral hepatic vein.31,44,52 One study noted that the left lateral and left medial hepatic veins entered the caudal vena cava separately after draining their respective lobes.31 Another study reported that the left lateral and medial lobar veins joined to form a main left hepatic vein before terminating on the caudal vena cava.44 The quadrate and right medial hepatic veins, which drain the central division of the liver, usually converge to form a single terminus approximately 1 cm in length (range, 0 to 3 cm) that subsequently joins the left hepatic vein.31 In some animals, the right medial and quadrate lobes may join the cava directly through a central hepatic vein or may drain into the left hepatic vein and through an accessory right medial liver lobar vein that enters the vena cava directly.43,44 The right lateral liver lobe is drained primarily by one large lobar vein that enters directly into the caudal vena cava. Some dogs may have one or two additional small branches draining this lobe. The lobar branch from the caudate process of the caudate lobe enters the cava 0.5 to 1.5 cm proximal to the entrance of the right lateral hepatic vein.31 At their terminations on the caudal vena cava, the right lateral and caudate hepatic veins are completely surrounded by hepatic parenchyma.15,182 The papillary process of the caudate lobe may have a separate opening if it has multiple branches or may drain into the left hepatic vein at the level of the bifurcation of the central and left divisions.44 The venous system of the abdominal cavity is derived from embryonic umbilical, vitelline, and cardinal veins (Figure 96-5).136 The right and left vitelline veins carry blood from the yolk sac to the sinus venosus, which forms the caudal part of the embryonic cardiac tube. The central segments of the vitelline vessels give rise to a venous plexus that forms the hepatic sinusoids. The cranial segment of the left vitelline vein atrophies, and the cranial segment on the right becomes the hepatic portion of the caudal vena cava. Two anastomoses develop between the caudal segments of the right and left vitelline veins. Portions of these anastomoses and their associated vitelline segments atrophy and reform to become the portal vein.121 Figure 96-5 Embryology of liver development. 1, Sinus venosus; 2, superior cardinal vein; 3, inferior cardinal vein; 4, left common cardinal vein. 5, right umbilical vein; 6, liver; 7, anastomosis between the left and right omphalomesenteric vein; 8, right umbilical vein (prehepatic); 9, left umbilical vein; 10, right omphalomesenteric vein; 11, umbilical vein (unpaired); 12, inferior vena cava; 13, ductus venosus; 14, portal vein; and 15, splenic vein. (Adapted from Schoni-Affolter F, Dubuis-Grieder C, Strauch E: Human embryology: an online course in embryology medical students. Swiss Virtual Campus, http://www.embryology.cn/indexen.html.) Paired umbilical veins, which carry blood from the allantois to the sinus venosus, subdivide into cranial, middle, and caudal segments as the liver develops. The cranial and right caudal segments atrophy, and the middle segments contribute to hepatic sinusoid formation. The left umbilical vein becomes the major conveyance of placental blood to the embryonic liver. A portion of this oxygenated blood flows through the hepatic sinusoids, but the majority flows directly to the heart via the ductus venosus, which is a venous shunt between the left umbilical vein and the cranial segment of the right vitelline vein.121,133 Three pairs of cardinal veins develop in the embryonic trunk: the caudal cardinal, subcardinal, and supracardinal veins. Portions of these vessels eventually degenerate and anastomose with each other and the cranial segment of the right vitelline vein to form the caudal vena cava within the trunk. The right and left supracardinal veins give rise to the abdominal segments of the azygos and hemiazygos veins, respectively.121,133 In a normal animal, the only residual communication between the embryonic cardinal and vitelline systems is the junction of the prehepatic and intrahepatic segments of the caudal vena cava.136 Developmental errors, however, can produce abnormal, functional connections between the cardinal and vitelline systems, resulting in congenital extrahepatic portocaval and portoazygos shunts.136 In some patients, the caudal vena cava may also be interrupted.55 Extrahepatic and right or central intrahepatic portocaval shunts may result from persistent connections between the caudal and right cranial segments of the vitelline system or malformation of hepatic sinusoids.105 The author (KMT) suspects that persistence of single congenital extrahepatic PSS shunts in the Yorkshire terrier and other toy breeds results from concurrent portal microvascular underdevelopment that increases intrahepatic portal resistance, forcing vestigial anomalous vessels to remain open. Numerous nonfunctional portocaval and portoazygos communications are normally present in fetuses; these may become functional if chronic portal hypertension develops, resulting in multiple acquired PSS.136 The umbilical vein and main (extrahepatic) portal vein terminate on the portal sinus, the vascular confluence of the right and left intrahepatic portal veins.119 In dogs, the ductus venosus arises from the portal sinus opposite the umbilical vein.171 After passing between the left lateral liver lobe and the ventral aspect of the papillary process of the caudate lobe, it terminates dorsally on the ampulla, an aneurysmal dilation at the confluence of the ductus venosus, left hepatic vein, and left phrenic vein.201,202 The anatomy of the cat ductus venosus is thought to be similar to that of dogs.200 Left-sided intrahepatic PSS most likely result from patency of the ductus venosus. In most dogs, the ductus venosus undergoes functional closure within 2 to 6 days after birth. Structural closure occurs within 3 weeks after birth, when connective tissue proliferation at the junction of the ductus venosus and portal sinus expands to the termination of the ductus at the left hepatic vein.102 Closure of the ductus venosus may be delayed in some breeds. In one study of Irish wolfhounds, the ductus venosus remained patent in 65% of puppies at 4 days of age and 23% at 6 days of age but was functionally closed in all puppies by day 9.102 The cause of ductus venosus closure is unknown, although hemodynamic factors, such as reduced umbilical venous flow, likely play an important role. In some species, the ductus venosus contains a functional ridge at its junction with the portal sinus. In near-term fetal lambs, smooth muscle within this ridge and along the vessel contracts in a dose-dependent manner in response to endothelin. Cytochrome P450 and thromboxane A2 also promote ductus contraction. Prostaglandin F1α and, to a lesser extent, prostaglandin E2 are natural constituents of the ductus venosus and cause relaxation of the vessel, modulating the effects of endothelin.1 In infants, prenatal administration of corticosteroids and postnatal administration of indomethacin decrease the time to ductus venosus closure.94 Corticosteroids inhibit phospholipase A2, decreasing prostaglandin production, and promote increase in activity of α-adrenoreceptors within the ductus venosus that also stimulate ductus venosus contraction.94 In newborn puppies, the ductus venosus appears to narrow uniformly after birth; no discrete anatomic sphincter is evident.28,171 Persistent flow through the ductus venosus could occur with increased sinusoidal pressure secondary to congenital portal hypoplasia28 or through imbalances of chemicals that stimulate or modulate the effects of endothelin in the ductus. The most recent classification suggests three separate categories of liver vascular disease: (1) congenital PSS; (2) disorders associated with abnormal hepatic blood flow or portal hypertension, currently termed primary hypoplasia of the portal vein (PVH); and (3) disturbances in portal outflow.13 The second category (PVH) includes processes that may or may not result in portal hypertension. These are termed PVH with portal hypertension and PVH without portal hypertension. PVH without portal hypertension was previously termed microvascular dysplasia (MVD); because microvascular dysplasia is a pathologic description associated with many conditions, the condition of PVH without portal hypertension will be referred to as PVH-MVD throughout this chapter. PSS can either be congenital or acquired in nature (Figure 96-6). Congenital shunting is reported in 0.18% of all dogs and 0.05% of mixed-breed dogs.179,180 Congenital PSS most commonly occurs as a single intrahepatic or extrahepatic vessel that provides direct vascular communication between the portal venous supply and the systemic venous circulation (caudal vena cava or azygous veins), bypassing the liver. Rarely, some animals have two or more congenital communications. There are several types of congenital PSS found in dogs and cats, including intrahepatic portocaval shunt, extrahepatic portocaval shunt, extrahepatic portoazygos shunt, portal vein atresia with resultant multiple portal-caval anastomoses, and hepatic arteriovenous malformations.* In dogs and cats, approximately 25% to 33% of congenital PSS are intrahepatic. Single extrahepatic shunts constitute 66% to 75% of congenital single PSS in dogs and cats, with a major solitary portocaval shunt being most common.12,167,174–176,210 Whereas most intrahepatic PSS are found in larger breed dogs, most extrahepatic PSS are seen in smaller breeds.210 Dogs with intrahepatic PSS generally have the largest volume of portal blood diverted through their shunt, allowing them to typically develop more severe clinical signs at an earlier age than those with extrahepatic PSS.12,75,147,167 Acquired shunts (20%) most commonly occur secondary to chronic portal hypertension; increased portal pressures result in opening of vestigial fetal blood vessels. Although usually a disease of older animals, acquired PSS have also been seen in young puppies. Acquired PSS vessels provide a means for handling the increase in hydrostatic pressure present within the portal system. They are usually multiple, tortuous, and extrahepatic in nature. Most connect a portal tributary directly to the renal vein or the caudal vena cava near the kidneys,18,25,39,40 but connections may occur with gonadal or internal thoracic vessels or other tributaries of systemic venous drainage. The most common causes of acquired extrahepatic shunts are hepatic fibrosis (cirrhosis), PVH with portal hypertension (congenital noncirrhotic portal hypertension),25 and hepatic arteriovenous malformationss.39 PVH with associated portal hypertension is a condition that has been termed idiopathic noncirrhotic portal hypertension.25,47 Other names include hepatoportal fibrosis, idiopathic hepatic fibrosis, veno-occlusive disease, idiopathic chronic liver disease, and nonfibrosing liver disease.81,142,145,187 The condition is characterized by intraabdominal portal hypertension, a patent portal vein, and a lack of cirrhosis on liver biopsy. The underlying cause of noncirrhotic portal hypertension is unknown; the speculated cause is a severe, diffuse intrahepatic microscopic vascular malformation. PVH-MVD without portal hypertension is a microscopic malformation of the hepatic vasculature previously called microvascular dysplasia (MVD).40,147 It is characterized by small intrahepatic portal vessels, portal endothelial hyperplasia, portal vein dilatation, random juvenile intralobular blood vessels, and central venous hypertrophy, often associated with some fibrosis. These lesions may allow for abnormal communications between the portal and systemic circulation at a microvascular level; however, the presence of true shunting has yet to be fully documented. PVH-MVD can occur as an isolated disease or in combination with macroscopic congenital PSS. In one textbook, 58% of dogs and 87% of cats with PVH had concurrent congenital macroscopic PSS.167 Clinical signs in dogs with PVH may be similar to those of PSS. However, when PVH exists without a macroscopic shunt, signs are often less severe, present later in life, and result in a better long-term prognosis with medical management alone. Breeds overrepresented are Cairn and Yorkshire terriers.40,147 No controlled studies have reported detectable differences in histologic changes in dogs with portal hypoperfusion secondary to congenital or experimentally induced PSS, portal hypoplasia without portal hypertension (PVH-MVD), or both conditions concurrently. Diagnosis of those conditions must therefore be based on results of additional imaging techniques and, in dogs with congenital PSS, response to surgery. Hepatic arteriovenous malformation is a rare condition in which multiple high-pressure arterial and low-pressure venous communications are present within the liver. This condition, previously termed hepatic arteriovenous fistula, is more appropriately named a malformation because most affected animals have numerous communications (malformations) instead of a single communication (fistula). Hepatic arteriovenous malformations have been described in dogs and cats and are usually congenital in nature.39,109 Typically, a branch of the hepatic artery communicates directly with the portal vein through numerous (tens to hundreds) aberrant shunting vessels within the liver, creating a high-pressure system that causes hepatofugal blood flow and arterialization of the portal vein. Because of resultant excessive portal hypertension, multiple extrahepatic shunts open to decompress the portal system. The long-term prognosis for patients with hepatic arteriovenous malformations is poorer than for animals with congenital portosystemic shunts, and early treatment should be considered.13,39,176 Hepatic encephalopathy is the cause of a majority of clinical signs seen in patients with PSS. It is a syndrome involving a gamut of neurologic abnormalities and occurs when more than 70% of liver function is lost.* The pathogenesis of hepatic encephalopathy is largely unknown and quite complex in human and veterinary medicine. A healthy liver filters a multitude of neurotoxic substances that are absorbed across the gastrointestinal barrier and drain through the portal system. The altered liver cannot appropriately perform its role in metabolism or substance clearance, permitting toxic substances to enter the systemic circulation through the portal–systemic communication(s). This results in effects on various organs; the central nervous system is most severely affected, resulting in an encephalopathic state. Multiple aspects of central nervous system metabolism have been implicated in the pathophysiology of hepatic encephalopathy. More than 20 different compounds have been found in excess in the circulation when liver function is impaired, including ammonia, aromatic amino acids, endogenous benzodiazepines, gamma-aminobutyric acid (GABA), glutamine, short-chain fatty acids, tryptophan, and others (Table 96-1).† These substances may impede neuronal and astrocyte function, causing cell swelling, inhibition of membrane pumps or ion channels, an elevation in intracellular calcium concentrations, depression of electrical activity, and interference with oxidative metabolism.85,86,155 These effects, in addition to an altered permeability of the blood-brain barrier seen in hepatic encephalopathy, impair cerebral function.33,85,86 Table • 96-1 Toxins Implicated in Hepatic Encephalopathy* *From references 8, 11, 12, and 23–25. From Berent A, Tobias K: Portosystemic vascular anomalies. Vet Clin North Am Small Anim 39:513, 2009. Ammonia is often considered the most important neurotoxic substance because increased concentrations trigger a sequence of metabolic events that have been implicated in hepatic encephalopathy in rats, humans, and dogs.10,33,72,85,86 Ammonia is produced by gastrointestinal flora and converted in the normal liver to urea and glutamine by the urea cycle. Ammonia is excitotoxic and associated with an increased release of glutamate, the major excitatory neurotransmitter of the brain. Overactivation of the glutamate receptors, mainly N-methyl-D-aspartate (NMDA) receptors, has been implicated as one of the causes of hepatic encephalopathy–induced seizures. With chronicity, inhibitory factors such as GABA and endogenous benzodiazepines surpass the excitatory stimulus, causing signs more suggestive of coma or central nervous system depression.10,85,86 Long-standing metabolic dysfunction, as seen with chronic liver impairment in PSS, results in alterations in neuronal responsiveness and energy requirements.33,86 Treatments that decrease ammonia concentrations, which are easily measured in animals, seem to reduce signs of hepatic encephalopathy. In humans, the degree of encephalopathy is not well correlated with the blood ammonia levels,54 suggesting that other suspected neurotoxins are also very important in pathophysiology of hepatic encephalopathy. Ammonia concentrations do not always correlate with signs of hepatic encephalopathy in veterinary patients, either. In fact, on rare occasions, dogs with normal ammonia concentrations can have obvious hepatic encephalopathy signs, but dogs with very high ammonia levels can seem neurologically normal. Coagulation abnormalities in patients with liver failure are multifactorial, depending on the interaction of the coagulation, anticoagulation, and fibrinolytic systems. Spontaneous hemorrhage is uncommon; hemorrhagic complications are usually induced with associated factors such as gastrointestinal ulceration, concurrent anticoagulation drugs (e.g., antiplatelet drugs, NSAIDs, steroid therapy), invasive procedures (surgery, aspiration or biopsy), or other concurrent medical problems. Suggested causes of coagulopathy in liver failure include decreased factor synthesis, increased factor utilization, increased fibrinolysis and tissue thromboplastin release, synthesis of abnormal coagulants (dysfibrinogenemia), decreased platelet function and numbers, vitamin K deficiency (particularly in bile duct obstruction), and increased production of anticoagulants.10,33 Extrahepatic PSS are most commonly seen in small- or toy-breed dogs such as Yorkshire terriers, Havanese, Maltese, Dandie Dinmont terriers, pugs, and miniature schnauzers. In a study of more than 2400 dogs in North America, these breeds were recently identified to have odds ratios of near 20 or greater for occurrence of a congenital PSS.117,179,210 In another study, Yorkshire terriers were reported to have an odds ratio for PSS of 35.9 times greater than that for all other breeds combined.174 A hereditary pattern has been documented in the Maltese dog and is thought to occur in Yorkshire terriers.175,176,178 In a study of Cairn terriers, the prevalence of congenital extrahepatic PSS was significantly higher than in the general population, and test matings of affected and unaffected related animals produced a higher prevalence of affected offspring.189 In that study, inheritance was considered autosomal and most likely polygenic or monogenic with variable expression. In cats, extrahepatic PSS are more commonly identified, although intrahepatic PSS are occasionally reported.14,150,176 PSS seems to be most commonly reported in domestic shorthairs, Persians, Siamese, Himalayans, and Burmese. 16,76,150,169,175 Intrahepatic PSS is overrepresented in larger breed dogs, with increased prevalence in Irish wolfhounds, retrievers (Labrador, golden), Australian cattle dogs, and Australian shepherds.95,175,176,195,211 Unusually, one study reported 71% of large-breed dogs had extrahepatic PSS; this was likely because of small numbers in the study.210 Left divisional intrahepatic PSS are considered heritable in Irish wolfhounds,90,128 and Australian cattle dogs and male dogs in Australia are overrepresented with right divisional intrahepatic PSS.95,170 Cairn terriers have been documented to have hereditary PVH-MVD that it is suspected to be an autosomal inherited trait, and Yorkshire terriers are also overrepresented with PVH-MVD, suggesting an inherited component to the condition.40,147 Animals with PVH with noncirrhotic portal hypertension are often purebred dogs, with Doberman Pinschers (27% of dogs) being overrepresented. Most dogs are younger than 4 years of age and weigh more than 10 kg.25 Animals with hepatic arteriovenous malformations can be any size and typically present at a very young age. A small number of cats have also been reported.11,39,117 Most dogs and cats with single congenital PSS present with signs of chronic or acute illness at a very young age (1 month to 2 years), although some animals have presented at 10 years of age or older and others shortly after birth.210,212 Multiple acquired extrahepatic shunts and PVH-MVD are usually diagnosed in older animals. In one study, the median age at diagnosis was 3 years (range, 7 months to 7 years) for dogs with multiple acquired PSS18 and 3.25 years (range, ≤10 years) in dogs with PVH.12,40 There is no clear gender predisposition for congenital PSS in dogs; males may be overrepresented in feline patients.167,210 Patient history in animals with single congenital PSS typically includes “failure to thrive” since birth; small stature (or the runt of the litter); a history of weight loss (11%) or failure to gain weight; intermittent episodes of dullness, lethargy, or “bizarre” behaviors (41% to 90%; including stargazing, head pressing, staring into walls or corners, random barking, intermittent blindness, pacing, or aggression); and a history of anesthetic intolerance.* Some animals present with a history of dysuria (20% to 53%).12,13,117 A complaint of polyuria and polydipsia is extremely common in dogs; potential causes include poor medullary concentration gradient because of low blood urea nitrogen (BUN) concentration, increased renal blood flow, increased adrenocorticotropic hormone (ACTH) secretion with associated hypercortisolism, and psychogenic polydipsia from concurrent hepatic encephalopathy.34,117,176 Abdominal effusion is seen in 75% of dogs with hepatic arteriovenous malformations; owners may complain of a “bloated” abdomen that may wax and wane. Abdominal effusion may also be seen with multiple acquired PSS and may be marked in patients with severe hypoalbuminemia and low oncotic pressure.39 The author ACB has noted that severe hypoalbuminemia is most common when animals have a concurrent protein-losing enteropathy, often associated with bleeding from gastrointestinal ulceration or inflammatory bowel disease with or without lymphangiectasia. This is more commonly seen in dogs with intrahepatic PSS compared to extrahepatic PSS. With PSS, central nervous system, gastrointestinal, and urinary systems are most commonly affected.12,117,176,210 Signs of hepatic encephalopathy can be either very obvious or quite subtle and are typically associated with abnormal behavior. More obvious central nervous system signs include lethargy, ataxia, unresponsiveness, pacing, circling, blindness, seizures, and coma.18,24,117,122,176 Correlation of onset of signs with meal ingestion has been reported in only 30% to 50% of patients. Gastrointestinal signs such as vomiting, diarrhea, anorexia, pica, and, gastrointestinal bleeding (melena, and hematemesis) occur in approximately 30% of dogs but are less frequent in cats.18,117,150,210 Preoperative gastrointestinal hemorrhage, although uncommon in dogs with extrahepatic PSS, may occur in 30% of large-breed dogs with intrahepatic PSS based on careful owner questioning.196,198 Preoperative treatment with antacids before shunt occlusion should therefore be considered in all dogs with intrahepatic PSS.9,11,196,198 Ptyalism is extremely common in cats (75%) and is thought to be a manifestation of hepatic encephalopathy or gastrointestinal upset.24,33,67,117,150 Many animals present with signs of lower urinary tract disease such as hematuria, stranguria, pollakiuria, or urinary obstruction (20% to 53%).11,12,210 Because of decreased urea production, increased renal ammonia excretion, and decreased uric acid metabolism, formation of ammonium urate calculi are common and can be associated with secondary bacterial urinary tract infections. In one study, urinary tract calculi were documented in 30% of patients with PSS.210 Animals with PSS may have other congenital defects concurrently. Cryptorchidism was reported in 30% of male cats in one study and 50% of male dogs in another.89,111 Additionally, heart murmurs can be found in both dogs and cats with PSS; these may be incidental flow murmurs in young patients or an indication of other congenital defects.24,111,117,176 Copper-colored irises inappropriate for the breed have also been documented with PSS, particularly in cats (Figure 96-7).37 Animals with PVH-MVD without macrovascular shunts have a similar signalment and clinical signs to those described above. However, these dogs and cats are typically older, and signs are frequently mild or nonexistent.40,117 Animals with PVH with portal hypertension show signs similar to animals with PSS and animals with hepatic cirrhosis with concurrent portal hypertension, such as ascites (60%), polyuria and polydipsia, gastrointestinal upset, hepatic encephalopathy, and weight loss. Clinical signs associated with hepatic arteriovenous malformations may be acute or chronic in nature. Signs are typically associated with portal hypertension and shunting through multiple extrahepatic PSS or from abdominal distention secondary to ascites. Gastrointestinal signs are common and can be very severe, and many dogs present with stunted growth and lethargy.39 Ascites is not seen in all affected animals; in one study, it was documented in 75% of affected dogs. Its absence was presumed to be from decompression of the portal system by the acquired shunts. Heart murmurs were documented in 20% of dogs with hepatic arteriovenous malformations in one study.39 Signs of hepatic encephalopathy are reported less frequently with hepatic arteriovenous malformations. In some animals, the sound of turbulent blood flow (bruit) over the fistula can be appreciated when the liver is auscultated. Microcytosis has been reported with or without associated normochromic, nonregenerative anemia in 60% to 72% of dogs and is documented in only 30% of cats.26,157,176 Microcytic anemia in animals with PSS may result from defective iron-transport mechanism, decreased serum iron concentrations, decreased total iron-binding capacity, or increased hepatic iron stores in Kupffer cells. These findings suggest that iron sequestration is the cause; however, a definitive cause is unknown.26,157 Typically, microcytosis resolves after shunt fixation and is not routinely seen in dogs with PVH-MVD.40,117 Morphologic changes in erythrocytes include target cells in dogs and poikilocytes in cats.208 Leukocytosis may occur and has been associated with a poor prognosis in some studies.12,167,195,210 Inadequate hepatic endotoxin and bacteria clearance from the portal circulation has been suggested as a possible explanation.177 Serum biochemical abnormalities are extremely common in dogs and cats with PSS. Deficiencies typically result from decreased hepatic synthesis and include hypoalbuminemia (50%), reduced BUN (70%), hypocholesterolemia, and hypoglycemia. In cats, hypoalbuminemia is uncommon, and low BUN concentrations are more frequently identified.67,150 Mild to moderate increases in serum liver enzyme activities (two- to threefold increases in alkaline phosphatase and alanine aminotransferase are also very common),208 and creatinine is often decreased. These abnormalities are typical of any hepatic vascular anomaly, and absolute levels of biochemical changes are not considered pathognomonic for any one of these particular conditions. Interestingly, alkaline phosphatase is typically higher than the alanine aminotransferase in dogs with PSS, likely from the contribution of bone isoenzyme in these growing animals. Hepatic organelle injury and increased release or decreased elimination of canalicular alkaline phosphatase has also been proposed.117,208 It is typically uncommon to see enzyme values more than fourfold the high end of the reference range. If this is seen, it is important to consider other concurrent underlying liver disease such as leptospirosis, cirrhosis, or chronic hepatitis. Liver biopsy at the time of shunt attenuation is recommended when liver enzymes are markedly increased. Common urinalysis abnormalities include decreased urine specific gravity and ammonium biurate crystalluria. More than 50% of affected animals are hyposthenuric or isosthenuric.24,117,161,176 Low urine specific gravity likely results from hepatic encephalopathy and associated psychogenic polydipsia and a poor medullary concentration gradient from the decreased urea production in the liver. Ammonium biurate crystalluria is reported in 26% to 57% of dogs and 16% to 42% of cats.42,89,188 Hyperammonuria from the deficient hepatic urea cycle combined with hyperuricacidemia from a deficiency in hepatic purine and pyrimidine metabolism (uric acid cycle) results in excessive ammonia and urate excretion by the kidneys. These compounds can precipitate into crystals or stones in the kidney or bladder; in one study, urinary tract calculi were noted in 30% of cases.33,161,210 Proteinuria is often seen in dogs with PSS and is suspected to be secondary to glomerular sclerosis or another underlying glomerulopathy. In one study of 12 dogs, 100% had evidence of a moderate to severe glomerulopathy characterized by glomerulofibrosis, membranoproliferative glomerulonephritis, or both.173 This link between severe liver disease and glomerulonephritis, termed cirrhotic glomerulosclerosis, has been seen in humans for many years and is speculated to be an immune-mediated glomerulonephritis from accumulation of antigens reaching the kidneys that would normally have been cleared by the liver.17 The test of choice for evaluating liver function in animals with PSS is measurement of fasting (12 hour) and 2-hour postprandial serum bile acids concentrations. Within the liver, bile acids are synthesized, conjugated, and secreted into the bile canaliculi, where they are collected and stored in the gallbladder until released into the duodenum after a meal. Bile acids aid lipid absorption via intestinal fat emulsification and metabolism. They are reabsorbed from the ileum, transported into the portal venous system, and extracted by hepatocytes for enterohepatic recirculation.35,36,38 Bile acids therefore reflect production, excretion, and recirculation and can be affected by the timing of the gallbladder contraction, rate of intestinal transport, degree of bile acid deconjugation in the small intestine, rate and efficiency of bile acid absorption in the ileum, portal blood flow, and functionality of hepatocyte uptake and canalicular transport. In animals with PSS, a persistent increase in bile acid concentrations results from shunting of reabsorbed bile acids into the systemic circulation.33,36,208 In some studies, increases in postprandial bile acids are 100% sensitive for detection of a PSS in dogs and cats; other studies have found that paired samples, but not individual measurements, are 100% sensitive.33,34,36,38 A small subset of animals has increased fasting and normal postprandial bile acids, and an even larger number of animals have normal fasting and increased postprandial bile acids.34,210 Normal Maltese dogs have been identified to have increased serum bile acids without other clinicopathologic evidence of hepatocellular dysfunction, possibly as a result of chemical interference with spectrophotometric analysis.170 Other “false”-positive results not specific to PSS occur with inappropriate sample timing, other hepatobiliary diseases, cholestasis, glucocorticoid or anticonvulsant therapy, tracheal collapse, seizures, and gastrointestinal disease.33,35,36,208 Falsely decreased results may occur with delayed intestinal absorption from prolonged transport time, lack of gallbladder contraction, inadequate food intake, delayed gastric emptying, and malabsorption or maldigestion. Spontaneous gallbladder contraction may occur between meals, resulting in preprandial bile acid concentrations that are greater than postprandial. When false-negative results from bile acid testing are suspected, a baseline ammonia evaluation or ammonia tolerance test can be performed. The primary source of blood ammonia is from the gastrointestinal tract, with more than 75% being generated by bacterial metabolism in the colon.24,32,33 Ammonia is delivered in the portal blood to hepatocytes, which convert it to urea via the urea cycle. In animals with PSS or other liver dysfunction, this conversion does not occur efficiently, resulting in increased serum ammonia concentrations. Baseline ammonia concentrations are not as sensitive as serum bile acid measurement; ammonia concentrations are abnormal in only 62% to 88% of animals with PSS, especially after prolonged fasting or with effective medical management of hepatic encephalopathy.36,41,97,170,210 False-positive (Irish wolfhound puppies) and false-negative results for baseline ammonia concentrations have been documented.125,127,128,194,210 In one study, measurement of 6-hour postprandial blood ammonia concentrations increased the sensitivity of the ammonia tolerance test to 91% in dogs with PSS.193 Animals with normal baseline ammonia concentrations and suspected liver disease can be challenged with administration of ammonia to determine their ability to clear the substance. The ammonia tolerance test can be performed by administration of ammonium chloride orally or rectally; the rectal route is better tolerated and easy to perform. Samples are evaluated before and 30 minutes after ammonium chloride administration (100 mg/kg [maximum, 3 g], administered as 2 mL/kg of a 5% solution in water) with a catheter placed deep rectally. Timing for semiquantitative analysis is at 20 and 40 minutes.35 Ammonium chloride can also be given via nasogastric tube or oral capsule, although the risk of vomiting and aspiration is higher with this route. Oral or nasogastric administration is contraindicated in animals with concurrent hepatic encephalopathy.125 Sensitivity of the ammonia tolerance test for hepatic insufficiency is 95% to 100%.125,143,161 Because the ammonia tolerance test gives semiquantitative values of the degree of portosystemic shunting, it has also been used to evaluate postoperative course after surgical attenuation of the shunt. It should be avoided, however, in animals in which the basal ammonia concentration is already increased or when increased serum bile acids and other imaging modalities support the diagnosis of PSS.35,159 Results of ammonia tests are affected by multiple variables, including sample handling. Plasma separation and laboratory analysis need to be performed within 20 minutes of sample collection, making this test difficult to perform in many practices. Prolonged fasting, a protein-restricted diet, and lactulose administration may decrease the basal ammonia concentration but does not affect ATT.58,62,144,208 False-positive test results in Irish wolfhound puppies have been documented because of an inborn error of ammonia metabolism in this breed.11,125,210 Severe hyperammonemia in the absence of PSS has been reported in cats with an inborn error in ammonia metabolism from a deficiency in the urea cycle enzyme ornithine transcarbamylase.194 Other causes of hyperammonemia in young animals include methylmalonic acidemia, other urea cycle enzyme deficiencies, and urethral obstruction–induced hyperammonemia.117,194 These conditions are not associated with PSS; therefore, hepatic biochemical and serum bile acid values are normal. Liver parenchymal cells synthesize most of the clotting factors (I, II, V, IX, X, XI, XII, XIII [and VIII via the liver vascular endothelium]). The liver is also involved in the regulation of coagulation by aiding in clearance of activated factors so that regeneration of inactivated factors and fibrinolytic factors can occur.7,96,140 The loss of approximately 65% to 80% of factors needs to occur before prolongation in prothrombin time (PT) or activated partial thromboplastin time (aPTT) results.33,96 Whereas animals with chronic liver disease typically have prolongations in aPTT alone, animals with acute liver disease often have prolongations in both PT and aPTT.140 Dogs with PSS frequently have coagulation abnormalities; however, spontaneous bleeding is very rare and does not usually occur until surgical intervention has been attempted.7,96,134 In one study, postoperative mortality increased in dogs that had a dramatic worsening of their coagulopathy after surgery.97 Frequently, dogs with PSS have prolonged preoperative aPTT, often without PT prolongation. Prolonged coagulation times in animals with PSS may include impaired hepatic synthesis, qualitative abnormalities, or abnormal clearance of coagulation factors.96,97,132 Interestingly, measured factor deficiencies in dogs with PSS involved the common (factors II, V, and X) and extrinsic pathways (factor VII) that did not necessarily result in PT prolongation. The precise reason for normal PT in the face of these factor deficiencies in PSS is not yet clear.96 In one study, platelet counts in dogs with PSS were lower than those of normal dogs before surgery. After surgery, the mean platelet count (161,000/µL) in dogs with PSS decreased 27% from baseline.96 Platelet counts were not low enough to cause clinical bleeding, however; therefore, postoperative decrease may have resulted from consumptive coagulopathy. Overall, coagulation status (PT, aPTT, and platelet counts) returned to normal by 6 weeks after successful PSS surgical fixation, but not in animals with persistent shunting.96 Protein C is a vitamin K–dependent serine protease enzyme that is activated by thrombin. Activated protein C provides antithrombotic, antiinflammatory, and antiapoptotic effects. In normal dogs, protein C activity is 70% or greater. Deficiencies in protein C can occur with decreased production (e.g., liver disease), consumption (e.g., disseminated intravascular coagulation [DIC], thrombotic episodes, surgery), renal disease, and malignancy. Measurement of protein C activity has been suggested for differentiation of PSS and PVH-MVD. In one study, 88% of dogs with PSS had protein C levels below 70%, and 95% of dogs with PVH-MVD had protein C levels 70% or above.184 Because protein C activity overlaps with a variety of liver diseases and because it does not differentiate normal dogs and dogs with PVH-MVD, it cannot be used as a sole discriminating test for the presence or type of liver disease. It may be helpful in trying to discern between portosystemic shunts and PVH-MVD. Effusion is rarely seen in animals with single congenital PSS alone unless they have severe hypoproteinemia from decreased hepatic production or severe gastrointestinal bleeding; more common causes in dogs with liver disease include portal hypertension associated with hepatic arteriovenous malformations, noncirrhotic portal hypertension, portal atresia, or acquired multiple extrahepatic PSS (chronic liver disease or cirrhosis). Typically, abdominal fluid for any of these conditions is a pure transudate that is clear and relatively acellular with a total protein below 2.5 (g/dL), specific gravity below 1.017, and less than 1000 nucleated cells/µL.208 The most common hepatocellular histopathologic findings reported in dogs with congenital PSS include bile duct proliferation, hypoplasia of intrahepatic portal tributaries, hepatocellular (lobular) atrophy, arteriolar proliferation or duplication, lipidosis and cytoplasmic vacuolar changes (lipogranulomas), smooth muscle hypertrophy, increased lymphatics around central veins, and Ito cell and Kupffer cell hypertrophy.6,33,84,135,162 Some animals have evidence of mild fibrosis around the central veins, and few have signs of necrosis or inflammation.33,135 Historically, histologic features associated with a poor prognosis included fibrosis, biliary hyperplasia, and necrosis.6,33,117 A recent paper, however, did not find any statistical association between histologic features and survival times of dogs with congenital extrahepatic or intrahepatic PSS.135 In another study, the association of lipogranulomas and prognosis was evaluated.135 Lipogranulomas are focal lesions consisting of Kupffer cells or macrophages that contain cytoplasmic brown (ceroid and hemosiderin) pigments and lipid vacuoles.84 Lipogranulomas have been reported in 55% of dogs with congenital PSS.84 Although they were significantly more common in dogs with PSS than age-matched control subjects, their presence was not associated with prognosis.135 Dogs with PVH without portal hypertension (PVH-MVD) and PVH with portal hypertension share similar hepatic histopathologic changes to those with macroscopic congenital shunts and can be misinterpreted by the pathologist if a proper medical history is not provided.25,40,47 Dogs with noncirrhotic portal hypertension often have more significant fibrosis extending into the parenchyma, particularly along the portal tracts or even bridging to other portal areas or central veins.25 In dogs with hepatic arteriovenous malformations, biopsies taken from liver lobes not involved in the arteriovenous communication often have similar histopathologic findings to those with PSS. Liver tissue in close proximity to the malformation often has largely dilated portal venules, marked arteriolar hyperplasia and muscular proliferation, and sinusoidal capillarization. Some portal veins can have evidence of thrombus formation and recanalization.39,117 Animals with hepatic encephalopathy can have histopathologic changes in the central nervous system, such as hypertrophy and hyperplasia of cerebral cortical protoplasmic astrocytes and polymicrocavitation of the brainstem, cerebellar nuclei, or cerebral cortex.161,176 Abnormalities on survey abdominal radiographs that are suggestive of but not diagnostic for PSS include microhepatica (60% to 100% of dogs and 50% of cats with PSS) and bilateral renomegaly.117,176 In dogs with PVH, the liver and kidneys can be normal in size radiographically.40,117 Ammonium urate or biurate calculi within the bladder, ureters, or kidneys are often radiolucent; however, those with additional calcium salt or struvite deposits may be radiopaque. To definitively diagnose a macroscopic shunt(s), additional imaging such as abdominal ultrasonography, scintigraphy, angiography (portal or arterial), computed tomographic angiography, or magnetic resonance angiography is needed. Ultrasonography is the most widely used tool for diagnosis of PSS in most practices. It is noninvasive, typically does not require anesthesia, and does not require special handling or licensing. Ultrasonography is also useful for detecting radiolucent uroliths in dogs and cats with hepatic vascular anomalies.101,103,104 Common findings on ultrasound in animals with congenital PSS include a decreased number of hepatic and portal veins, a subjectively small liver, and an anomalous vessel (Figure 96-8). Extrahepatic shunts are more difficult to diagnose with ultrasonography than intrahepatic PSS because of small patient size, small vessel size, variable PSS location, presence of gas in the bowel and lungs obscuring the image, and activity of the patient during scanning. This is why many sonographers prefer animals to be sedated if a shunt is expected to improve the chances of appropriate and accurate diagnosis. Care should be taken to evaluate for a portophrenic shunt, which is often found cranial to the liver and terminates, via the left phrenic vein, on the caudal vena cava at the level of the diaphragm. Air in the lungs can make this shunt difficult to find. Multiple extrahepatic shunts are even harder to find and are typically located near the left kidney. There is considerable variation in reported accuracy of ultrasonography for detection of shunts, with sensitivities ranging from 74% to 95% and specificity from 67% to 100%.73,101,103–105,168 Correct distinction of intrahepatic PSS from extrahepatic PSS was possible in 92% of cases in one study.101 The overall sensitivity was higher for detection of intrahepatic PSS (95% to 100%) compared with extrahepatic PSS because of the presence of the liver parenchyma surrounding the shunting vessel and typically large size of the shunting vessel, which make visualization easier.101 In general, the sensitivity and specificity of ultrasonography for detection of PSS is operator and experience dependent. If the PSS cannot be identified, color-flow and pulse-wave Doppler imaging may detect changes in flow direction. Classically, hepatic arteriovenous malformations have hepatofugal flow, and extrahepatic PSS have hepatopedal flow through the portal vein (Figure 96-9). In normal dogs, portal flow velocity was estimated to be 15 cm/sec, with a uniform velocity and direction.117 Flow velocity was increased or variable in 53% of dogs with extrahepatic PSS and in 92% of dogs with intrahepatic PSS.117 Dogs and cats with extrahepatic portosystemic shunts have also been documented to have a reduced portal vein : aorta size. Another useful, noninvasive method for detecting a PSS is nuclear scintigraphy. Technetium pertechnetate (99mTc pertechnetate) is the radioisotope most commonly used for this procedure.57,103 For transcolonic scintigraphy, a bolus of the isotope is infused per rectum into the colon, and the animal is imaged using a gamma camera. The isotope is absorbed and drained through the colonic veins that empty into the caudal mesenteric vein and ultimately the portal vein. In a normal patient, the radioisotope first travels through the liver before reaching the heart; the heart is visible on imaging 8 to 14 seconds after injection.103 In a patient with a PSS, the isotope travels from the portal vein to the heart, bypassing the liver and then returning to the liver through the hepatic arterial circulation. Therefore, radioactivity in the heart arrives sooner and in greater concentration than expected. Administration of 99mTcO4− radionuclide through a transsplenic injection is becoming more common at specialty centers.130 With transsplenic administration, a dose of radionuclide that is 10% to 25% that of transcolonic administration is injected under ultrasound guidance into an intraparenchymal splenic vein. Animals are usually lightly anesthetized with propofol to facilitate injection and restraint for imaging. If a PSS is present, radioisotope often reaches the heart within 2 to 4 seconds after administration (Figure 96-10). In one study, transsplenic scintigraphy was 100% sensitive and specific for diagnosis of congenital PSS and significantly more likely than per rectal administration to detect shunt number and termination (azygos vein or caudal vena cava). Clearance times are faster with transsplenic scintigraphy, and a smaller amount of radionuclide is used, reducing exposure to personnel.160 A shunt fraction can be calculated from scintigraphic results to provide an estimate of the percentage of portal blood bypassing the liver. On transcolonic scintigraphy, a shunt fraction below 15% is considered normal; the shunt fraction is even lower in healthy animals undergoing transsplenic scintigraphy. The shunt fraction is normal in dogs with PVH-MVD if no acquired shunts are present and increased in dogs and cats with single congenital intrahepatic or extrahepatic PSS, multiple acquired PSS, or hepatic arteriovenous malformations with acquired PSS. Results of scintigraphy cannot be used to differentiate intrahepatic from extrahepatic PSS or normal dogs from dogs with PVH-MVD and do not consistently differentiate single from multiple shunts. Most dogs with congenital PSS have fractions above 60% to 80%; however, some cats have lower fractions than dogs (52% in one study).46 Because of variability in calculation of shunt fractions, scintigraphy has not proven useful for assessment of postoperative prognosis.146 In addition, if the material is administered too aborally in the rectum during transcolonic scintigraphy, absorption directly into the caudal vena cava occurs, resulting in falsely increased shunt fractions, decreasing test specificity. Uptake of radionuclide into the portal system may be poor in patients with portal hypertension that are undergoing transsplenic radioisotope injection. Computed tomographic angiography is the gold standard for the evaluation of the portal venous system in humans.69 It is noninvasive, fast, and provides imaging of all portal tributaries and branches with a peripheral contrast injection (Figure 96-11). This modality can be performed in dogs and cats of any size with accuracy when performed correctly, and it allows for further manipulation of the images after the study is complete. Dual-phase computed tomographic angiography provides a complete evaluation of portal and hepatic vasculature and is considered superior to single-phase computed tomography.217 Computed tomographic angiography is valuable for preprocedural planning in animals with suspected hepatic arteriovenous malformations or single or multiple intrahepatic PSS. Additionally, it can be used to detect single congenital PSS that are not seen on ultrasound, exploratory laparotomy, or intraoperative mesenteric portography. Direct injection into the splenic vein (transsplenic CT portography) provides more intense enhancement of splenic and portal veins with less contrast medium but causes inconsistent opacification of intrahepatic portal branches and parenchyma because of streamlining and streak artifacts.49 It also increases radiation exposure of personnel. As with computed tomographic angiography, magnetic resonance angiography provides a three-dimensional, preoperative image of the shunt to facilitate preprocedural planning (Figure 96-12). In the authors’ experience (ACB), images from dual-phase computed tomographic angiography are relatively easy to interpret, provide superior detail (particularly with newer multisliced CT scanners), are faster to perform, and are less costly than magnetic resonance angiography. Although the specificity is reported to be as high as 97%, the sensitivity of magnetic resonance imaging (MRI), without the use of gadolinium-enhanced angiography, is low (63% to 79%).152 The sensitivity will likely increase as the technique is improved. Operative mesenteric portography provides excellent imaging and localization of shunting vessels. Most commonly, it is performed at the time of laparotomy with a portable fluoroscope (C-arm). Alternatively, a jejunal or splenic vein can be catheterized and the abdomen closed so the patient can be transported to a radiology suite with a standing fluoroscopy unit. Approximately 2 to 4 mL/kg of water-soluble, sterile radiopaque contrast material (iohexol 240 to 360 mg/mL) is injected as a bolus, and fluoroscopy or radiography is used to image portal blood flow (Figure 96-13). Classically, differentiation of intrahepatic from extrahepatic PSS on portography is based on the point where the shunt diverges from the portal vein. If this point is cranial to the thirteenth thoracic vertebra, the shunt is typically diagnosed as intrahepatic; if it is caudal to the thirteenth thoracic vertebra, it is usually extrahepatic.14,176 Because many portoazygos and portophrenic shunts may terminate at the level of the diaphragm or esophagus, differentiation of intrahepatic and extrahepatic PSS based on point of divergence is not always possible. Sensitivity of intraoperative portography has been reported to be 85%, 91%, and 100% in dorsal, right lateral, and left lateral recumbency, respectively; imaging is improved with use of digital subtraction.11,117,175,176 Although rare, caudally located extrahepatic PSS may not be visible with transsplenic injection techniques if the portal tributary comprising the shunt is located upstream from the splenic vein.49 In most patients, however, these caudal shunts will opacify because of retrograde (hepatofugal) flow through the portal vein (see Figure 96-13). Splenic injection may result in life-threatening hemorrhage in patients with concurrent hepatic arteriovenous malformations if the splenic vein is arteriolized or in animals with severe portal hypertension. Other methods for contrast radiography of shunts include percutaneous ultrasound-guided splenic venography (see Figure 96-13), retrograde transjugular portography, and cranial mesenteric arteriography via the femoral artery. Cranial mesenteric arteriography requires a large volume of contrast material and is often very difficult to interpret, resulting in a lack of details one would obtain from computed tomographic angiography or other methods of portography. Medical management (Table 96-2) is recommended before any anesthesia performed for diagnosis or treatment (surgical or interventional radiology of congenital PSS, PVH-MVD, or noncirrhotic portal hypertension. Medical management should also always be considered for long-term therapy when a macrovascular shunt is detected but surgery is not possible or declined. Medical management controls clinical signs associated with shunting but does not resolve the underlying diminished hepatic perfusion; therefore, surgery is recommended in most patients with correctable disease. Table • 96-2 Medical Management for Portosystemic Shunts From Berent A, Tobias K: Portosystemic vascular anomalies. Vet Clin North Am Small Anim 39:513, 2009. When a patient presents with signs of hepatic encephalopathy, aggressive efforts should be instituted to stabilize the patient and decrease the ammonia concentrations. Nothing should be given by mouth until the patient is alert, aware, and able to safely swallow. If an animal presents recumbent and is unable to drink, or is dehydrated from gastrointestinal fluid loss, intravenous fluid therapy is administered to normalize and maintain hydration. Some clinicians avoid using lactated Ringer’s solution because of the need for hepatic conversion of lactate to bicarbonate; however, this issue is likely more theoretical than clinical. Hypokalemia may also contribute to hepatic encephalopathy, and patients with chronic diarrhea or inappetence may require potassium supplementation.101,124 Metabolic acidosis may also contribute to hepatic encephalopathy and should be corrected slowly with fluid therapy; rarely, sodium bicarbonate therapy is required. It is important to verify that concurrent respiratory acidosis does not exist before sodium bicarbonate therapy is begun and that ventilation is appropriate or assisted, if needed. Glucose should be supplemented in intravenous fluids, particularly in young puppies with PSS, in which glycogen stores and gluconeogenesis are minimal. Therapy for acute, severe hepatic encephalopathy includes administration of warm water enemas, oral or rectal lactulose, antibiotics to decrease urease-producing bacteria (metronidazole, ampicillin, or neomycin), and anticonvulsant therapy, if necessary. GABA and its receptors have been implicated in the pathogenesis of hepatic encephalopathy.11,12,33 In humans, administration of a benzodiazepine antagonist such as flumazenil (0.02 mg/kg IV to effect) may reverse hepatic encephalopathy–induced comas. Similar studies in veterinary patients have not been performed. Mannitol therapy is often considered because of a clear association between hepatic encephalopathy and cerebral edema in humans with severe hepatic encephalopathy or after significant seizure activity.10,12,33 Seizures not caused by hypoglycemia or hyperammonemia are initially treated with a benzodiazepine. Some clinicians prefer midazolam over intravenous diazepam, which contains a propylene glycol carrying agent that requires liver metabolism. Others feel that diazepam is the anticonvulsant of choice for immediate effect in an animal having seizures from hepatic encephalopathy associated with PSS. Diazepam is not recommended for animals with hepatic encephalopathy for other cause of liver failure (i.e., hepatic lipidosis). After seizures are controlled, loading doses of and continued treatment with phenobarbital, potassium bromide, sodium bromide, or levetiracetam (Keppra) may be considered (see Table 96-2), particularly if continued seizure activity is anticipated. At this point, there is little literature to support the use of levetiracetam in veterinary medicine to control hepatic encephalopathy–associated seizures, although this medication is preferred by many neurologists and is commonly used in the author’s practice (ACB). Levetiracetam can be used in an intravenous form, but therapeutic levels to be reached with a loading dose are unknown for this drug. It may also lose effectiveness after 6 months of administration.192 Other options include zonisamide (10 mg/kg BID), which is more convenient than thrice-daily administration of levetiracetam. Nutritional management is particularly important in young animals and any that have extremely poor body condition. The diet should be readily digestible, contain a protein source of high biologic value, supply enough essential fatty acids, maintain palatability, restrict components that precipitate hepatic encephalopathy (e.g., manganese), supply components that improve urea cycle or liver function (e.g., zinc and vitamin E, respectively), and meet minimum requirements for vitamins and minerals. Milk and vegetable proteins are lower in aromatic amino acids (tyrosine and phenylalanine) and higher in branched-chain amino acids (e.g., valine, leucine, isoleucine) than animal proteins. These sources are less likely to precipitate hepatic encephalopathy.10,12 Protein should be moderately restricted, with a dietary goal of 18% to 22% for dogs and 30% to 35% for cats (on a dry matter basis). In one study, use of soy- or meat-based moderately low-protein diets resulted in significant improvement in hepatic encephalopathy scores. Dogs fed soy-based protein diets had greater improvements in plasma ammonia concentrations and coagulation parameters than those fed the meat-based protein-restricted diets.141 Animals with intrahepatic PSS have a predisposition to the development of gastrointestinal ulcerations.11,110,196,197 Gastric bleeding and ulceration are treated with an acid receptor blocker, like famotidine or omeprazole, and sucralfate. Lifelong treatment with a proton pump inhibitor is currently recommended (ACB) in dogs that have undergone intrahepatic PSS attenuation. NSAIDs, steroids, and other potentially ulcerogenic medications should be used with caution in any dog with liver disease, particularly those with intrahepatic PSS. Nutraceuticals for hepatic supportive therapy have been recommended for a variety of liver diseases.56 Unfortunately, no controlled studies have evaluated their effectiveness for treatment of animals with hepatic vascular malformations. Options include S-adenosyl-L-methionine (SAMe), ursodeoxycholic acid, vitamin E, and milk thistle (silymarin). SAMe has hepatoprotective, antioxidant, and antiinflammatory properties, aiding in hepatocellular membrane structure, function, and fluidity. It also aids in metabolism of glutathione, which participates in many metabolic processes and plays a critical role in detoxification mechanisms of cells. Depletion of hepatic glutathione can indirectly cause toxic cellular effects by increasing oxidative stress, particularly in cats. Vitamin E, another antioxidant, prevents or minimizes lipid peroxidation within the hepatocytes. Silymarin is the active extract in milk thistle, which has demonstrated antioxidant and free radical scavenging properties in vivo and in vitro.10 Specifically, silymarin inhibits lipid peroxidation of hepatocyte and microsomal membranes and protects against gene damage by suppression of hydrogen peroxide, superoxide, and lipoxygenase. Silymarin also increases hepatic glutathione content, appears to retard hepatic collagen formation, and has hepatoprotective effects through inhibition of Kupffer cell function. Silymarin has an extremely low toxicity and has been used extensively in clinical patients with little concern for side effects. Few studies have evaluated the prognosis for animals with congenital PSS that are treated with medical management alone.53,63,195 One study reported that medically treated dogs developed progressive portal fibrosis.53 In another study of 27 dogs, 9 had extrahepatic PSS, 17 had intrahepatic PSS, and in one the shunt was complex.195 The median survival time of all dogs from time of diagnosis to euthanasia was 9.9 months, with an overall median age of 20 months at the time of euthanasia. Of the 27 dogs in the study, 14 (51.8%) were euthanized with a median survival time of 9.9 months, and 4 (14.8%) were lost to follow-up. One third (9 of 27) of the dogs survived long term (median survival time; 56.9 months; range, 5 months to >7 years), with many of those still alive at the time of follow-up.195 Outcomes varied with shunt location. Dogs with intrahepatic PSS on medical management alone had persistent or progressive neurologic and urinary tract signs, but gastrointestinal signs were less severe after medical management was initiated. In contrast, dogs with extrahepatic PSS had neurologic, gastrointestinal, and urinary clinical signs that occurred with similar or less frequency after they had been medically managed. Eleven of 17 (64.7%) dogs with intrahepatic PSS were euthanized, primarily because of uncontrolled signs of hepatic encephalopathy, and three of nine (33%) dogs with extrahepatic PSS were euthanized.195 Dogs treated medically had a significant decrease in total protein, alkaline phosphatase, and alanine aminotransferase concentrations over time; however, bile acids, BUN, albumin, and mean corpuscular volume did not change significantly. The prognosis was better in dogs that were older at the time of onset of clinical signs, had extrahepatic PSS, or had higher BUN concentrations. There were no correlations between survival times and bile acid, serum protein, albumin, alkaline phosphatase, or alanine aminotransferase concentrations or mean corpuscular volume.195 In another study, long-term survival was noted in 14 of 27 dogs (51.9%) managed medically and 87 of 99 dogs (87.9%) managed surgically.63 In that study, age did not have any significant effect on survival time. The prognosis is excellent for most dogs with PVH-MVD. In one study, 22 of 24 (92%) had good long-term survival or died of reasons unrelated to their liver disease.40 Of dogs with noncirrhotic portal hypertension, 13 of 33 survived long term (40%).25 Many of these dogs were euthanized because of intraoperative findings, however, and medical management was not attempted. The overall prognosis should be considered favorable.
Hepatic Vascular Anomalies
Anatomy
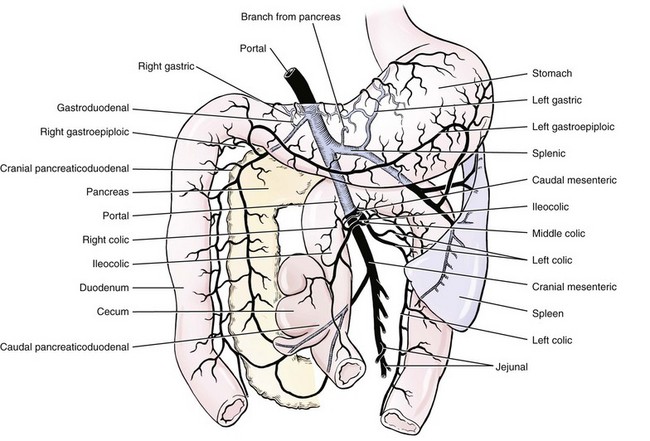
Portal Vein
Hepatic Artery
Hepatic Veins
Embryology
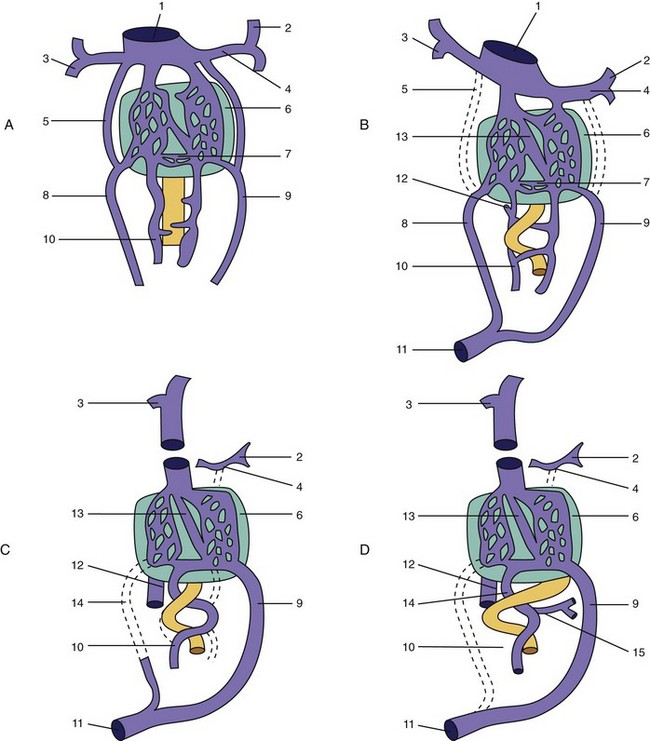
Portocaval and Portoazygos Shunts
Patent Ductus Venosus
Classification And Epidemiology
Portosystemic Shunts
Congenital Portosystemic Shunts
Acquired Portosystemic Shunts
Portal Vein Hypoplasia
Hepatic Arteriovenous Malformations
Pathophysiology
TOXINS
MECHANISMS
Ammonia
Increases brain tryptophan and glutamine; decreases ATP availability; increases neuronal and cellular excitability; increases glycolysis; can cause brain edema; decreases microsomal Na+,K+-ATPase in the brain
Aromatic amino acids
Decrease DOPA neurotransmitter synthesis; alter neuroreceptors; increase production of false neurotransmitters
Bile acids
Membranocytolytic effects alter cell membrane permeability; make the blood-brain barrier more permeable to other hepatic encephalopathic toxins; impair cellular metabolism because of cytotoxicity
Decreased α-ketoglutaramate
Diversion from Krebs cycle for ammonia detoxification; decreased ATP availability
Endogenous benzodiazepines
Neural inhibition through hyperpolarization of neuronal membrane
False neurotransmitters
Impair norepinephrine action
Tyrosine → Octopamine
Impair norepinephrine action
Phenylalanine → Phenylethylamine
Synergistic with ammonia and SCFA
Methionine → Mercaptans
Decreases ammonia detoxification in the brain urea cycle; gastrointestinal tract derived (fetor hepaticus breath odor in hepatic encephalopathy); decreases microsomal Na+,K+-ATPase
GABA
Neural inhibition by hyperpolarizing neuronal membrane; increases blood-brain barrier permeability to GABA
Glutamine
Alters blood-brain barrier amino acid transport
Phenol (from phenylalanine and tyrosine)
Synergistic with other toxins; decreases cellular enzymes; neurotoxic and hepatotoxic
SCFAs
Decrease microsomal Na+,K+-ATPase in brain; uncouple oxidative phosphorylation; impair oxygen utilization; displace tryptophan from albumin, increasing free tryptophan
Tryptophan
Directly neurotoxic; increases serotonin through neuroinhibition
Coagulation Disorders
Diagnostic Evaluation
History
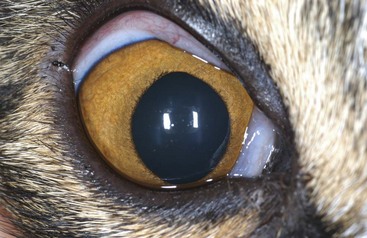
Clinical Signs and Examination Findings
Portal Hypoplasia
Hepatic Arteriovenous Malformations
Clinical Diagnosis
Liver Function Testing
Bile Acids
Ammonia
Coagulation Profiles
Protein C
Abdominal Effusion Evaluation
Histopathology
Diagnostic Imaging
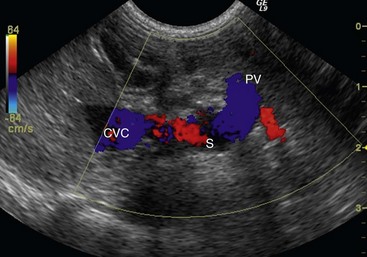
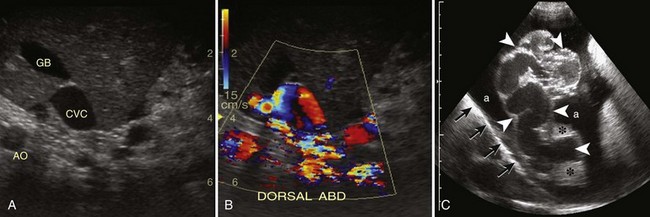
Abdominal Ultrasonography
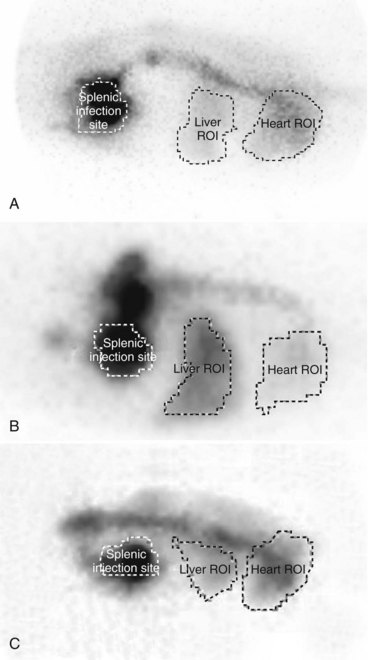
Scintigraphy
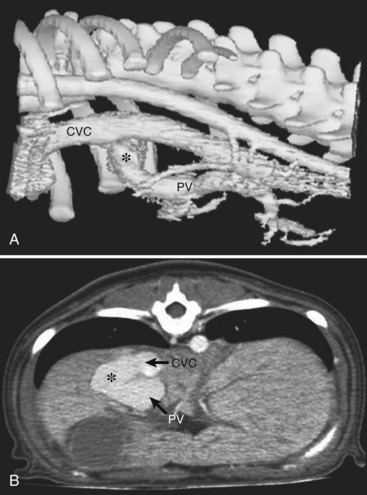
Computed Tomographic Angiography
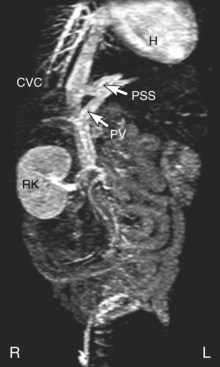
Magnetic Resonance Angiography
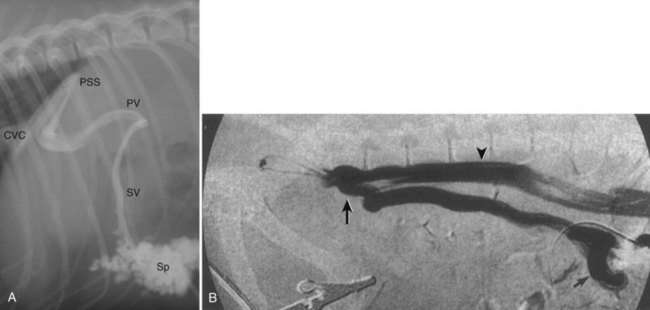
Portovenography
Treatment
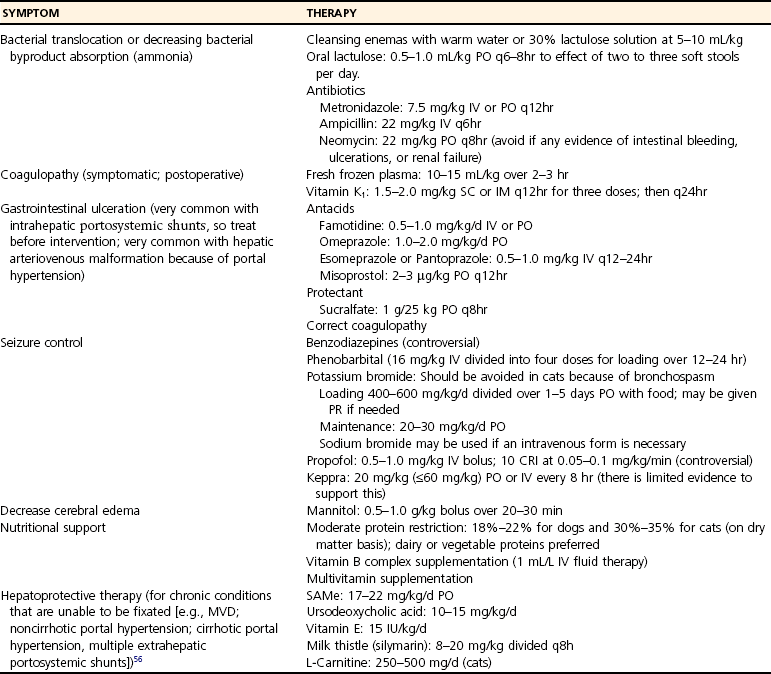
Prognosis With Medical Management Alone
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Hepatic Vascular Anomalies
Only gold members can continue reading. Log In or Register to continue
