Chapter 2 Gastrointestinal Microbiota
The intestinal microbiota is the collection of all live microorganisms that inhabit the gastrointestinal (GI) tract. The word microflora is often used synonymously, but microbiota (from bios, Greek: life) is the technically correct term. The intestinal microbiota plays an important role in GI health and disease, yet our understanding of the composition, dynamics, and functionality of the intestinal ecosystem remains rudimentary. The total microbial load in the intestine is estimated to be 1012 to 1014 organisms, approximately 10 times the number of body cells. It is estimated that the intestine harbors several thousand bacterial strains.1,2 This mutually interacting system comprising the host cells and the resident microbes is termed the intestinal microbiome. The microbiota can be influenced by exogenous factors such as diet and antibiotic administration, but it is usually resilient to these changes and returns rapidly to its pretreatment state. Therefore long-term treatment strategies for modulating the microbiota are necessary. New molecular tools have improved our understanding of microbial diversity in the intestine. Although the major phylogenetic lineages are similar, the microbiota differs substantially at the level of species and strain in each individual animal of the same species. Yet despite these differences, the metabolic end products in the intestine are very similar between individuals. New metagenomic approaches suggest the presence of a “core microbiome,” where the function of the intestinal ecosystem is independent of the presence of specific bacterial species or strains. For better understanding of microbial–host interactions in health and disease, future work must focus on the intestinal microbiome as one entity, evaluating its phylogenetic composition as well as metabolic functions.
Methods for Characterization of the Intestinal Microbiota
Practical Considerations
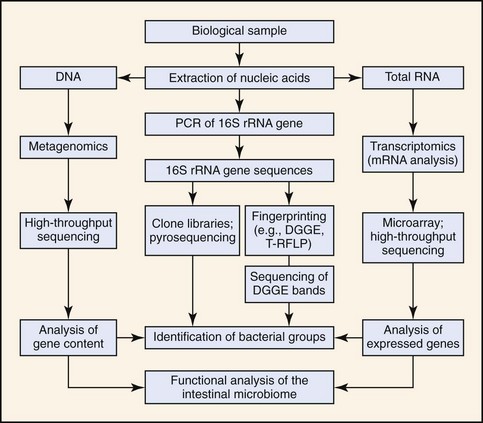
Methods for characterization of the intestinal microbiota are based on cultivation techniques or molecular tools (Fig. 2-1). The selection of the best approach depends on the study problem (e.g., detection of specific pathogens in clinical specimens, or general characterization of the intestinal ecosystem), the cost, and the availability of technologies. Each method has strengths but also limitations (outlined below). For general ecologic surveys of microbial communities, molecular high-throughput sequencing techniques yield the most information as they allow in-depth identification of microorganisms. For screening of specific pathogens, culture techniques and species specific polymerase chain reaction (PCR) assays may be most useful. Both of these methods are sensitive to sample handling and processing. Detailed instructions for sample collection and shipping for each particular assay should be acquired before sample submission as many laboratories use their own in-house culture or PCR assays. In the case of molecular methods, there is usually no standardization of DNA extraction or PCR protocols among laboratories, and such factors impact on the sensitivity and specificity of the assays. Improper DNA extraction, especially from fecal samples, may result in the presence of residual PCR inhibitors that cause false negative results. Because of the high sensitivity of PCR assays (theoretically a single target copy can be amplified), any DNA contamination can lead to false-positive results. A laboratory should be chosen that has expertise in molecular analysis, and that has validated each assay in the target specimen.
Bacterial Culture
Several limitations are associated with culture methods, especially if they are used to survey for the presence of unknown microorganisms in intestinal samples. Bacterial culture underestimates total bacterial numbers in the intestine, as microscopic counts (especially when using fluorescent dyes) are typically higher than the total viable counts obtained from culture. Although the majority of intestinal bacteria cannot be cultured, this does not necessarily mean that they are uncultivable, but rather, that insufficient information is currently available about their optimal growth requirements. Furthermore, many microbes depend on mutualistic interactions with other bacteria and the host, hindering their successful isolation in vitro. While recent advances have increased the cultivable fraction,3 it is estimated that less than 10% of intestinal bacteria can be cultured, and an even smaller fraction can be correctly classified. Therefore studies of the intestinal ecosystem may exhibit bias toward the minor cultivable portion of the gut microbiota. The analytical sensitivity of bacterial culture depends on the organism and its growth requirements. Bacterial culture is associated with difficulties in handling, storing, and shipping of clinical specimens. Ideally, samples should be processed immediately to preserve anaerobic species. Many selective culture media lack sufficient specificity, and often organisms other than the target are enumerated.4 Phenotypic and biochemical identification systems often fail to accurately classify many microorganisms, requiring DNA sequencing of isolates.
Molecular Techniques
Because bacterial culture underestimates microbial diversity, the use of molecular tools has now become the standard approach in microbial ecology.5–7 The principle of these methods is that DNA or RNA is extracted from intestinal samples, and a specific gene is amplified with universal primers that target conserved regions (located up- and downstream of variable regions within the gene).8 This approach allows in theory the amplification of DNA from all known and unknown bacterial species in a sample (see Fig. 2-1). The mixture of sequences can then be separated by subcloning and identified by sequencing, or they can be separated by methods that yield a “fingerprint” of the bacterial community.8,9 The 16S ribosomal RNA (rRNA) gene is most commonly targeted as more than 1.6 million unique sequences are available in public databases (Ribosomal Database Project; http://rdp.cme.msu.edu/). Other more rarely used genomic targets include the 16S to 23S internal transcribed spacer (ITS) region or the chaperonin (cpn60) sequences.10 If the sequence for a particular phylotype is known, specific PCR assays can be designed for its detection. Real-time PCR assays (with universal-, group-, or species-specific primers) can be used for quantitative analysis. Novel techniques analyze total genomic DNA or messenger RNA (mRNA) without prior amplification of specific genes and yield information about the gene content (metagenomics) or the expressed genes (transcriptomics) of the intestinal microbiome.
Molecular Fingerprinting
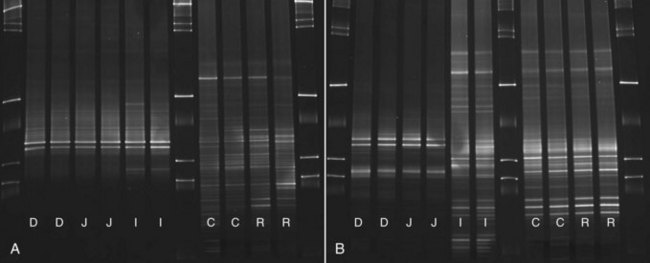
Molecular fingerprinting techniques are used for simultaneous analysis and comparison of microbial communities in multiple samples. These techniques provide information on microbial changes over time and in response to treatment. Available techniques include denaturing gradient gel electrophoresis (DGGE), temperature gradient gel electrophoresis (TGGE), and terminal restriction fragment length polymorphism (T-RFLP). The goal is to separate the mixture of PCR amplicons that were generated with broad range primers (universal or group specific) to yield a “fingerprint” of the bacterial community. This is achieved as each bacterial phylotype has a unique nucleotide composition (i.e., guanine+cytosine content). These differences in nucleotide composition result in unique melting behaviors of PCR amplicons. Each PCR amplicon reaches a specific melting point in a different position in a polyacrylamide gel, where it will denature and slow its migration. This banding pattern illustrates the bacterial diversity in the sample (Fig. 2-2). In DGGE, a gel containing a linear gradient of DNA denaturants is used, whereas in TGGE a temperature gradient is used for separation. Bands of interest can be excised and sequenced. DGGE and TGGE are inexpensive and rapidly performed. However, because DGGE/TGGE bands are usually short, only limited resolution of PCR amplicons can be achieved, and many bacterial phylotypes will have similar or the same melting behaviors. Therefore, these techniques yield typically only 20 to 40 bands, capturing only changes in the predominant bacterial groups. The use of T-RFLP allows profiling, but also quantification of microbial communities. Bacteria are amplified in PCR assays containing a fluorescent labeled primer. The PCR products are then fragmented by size with sequence specific restriction enzymes. The fragments are separated by capillary electrophoresis with subsequent quantitative measurement of the fluorescence.
Identification of Bacterial Groups
The amplification of the 16S rRNA gene with universal primers that target conserved regions allows amplification of theoretically all bacteria present in the sample. For identification of individual bacterial phylotypes, PCR amplicons must be separated and sequenced. A commonly used method is the construction of 16S rRNA gene clone libraries.6,7,10 The PCR amplicons are separated by ligation into plasmid vectors with subsequent transformation into Escherichia coli cells. These cells are plated on culture medium and grown overnight. Each cell forms a colony containing one plasmid with the original amplified 16S rRNA gene sequence. This plasmid can then be purified and sequenced. Although this approach is informative, it is laborious and not well suited for analysis of large sample numbers. Recently, new high-throughput sequencing platforms have been introduced that allow automated separation of PCR amplicons without the need for subcloning. These platforms (e.g., 454-pyrosequencing, Illumina) allow several thousand sequences to be analyzed within a few hours, yielding a deep coverage of the microbiota.2,11 However, because of the high bacterial diversity in the intestine, groups of low abundance (especially pathogens of interest) may constitute such a low proportion of the total bacteria, that they still escape identification. Therefore, for the detection of particular groups of interest (i.e., Bifidobacterium spp.), the use of group specific PCR primers is recommended.
Techniques based on the 16S rRNA gene also have limitations. Bias is inherent during DNA extraction, primer selection, PCR amplification, and sequence analysis. Some commonly used primers and PCR protocols underestimate the presence of specific bacterial groups, especially those with a high guanine+cytosine content (e.g., Bifidobacterium spp.),7 and some researchers use either a primer mix or group-specific primers for more accurate amplification.11
Quantification of Bacterial Groups
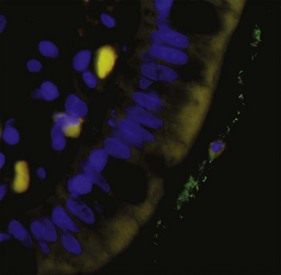
Commonly used methods include quantitative real-time PCR assays,10,12 fluorescent in-situ hybridization (FISH),13 RNA dot blot hybridization,14,15 and flow cytometry of fluorescent-labeled probes. Universal-, group-, or species-specific primers can be utilized. FISH allows for quantification of bacterial groups (Fig. 2-3), and this method also permits visualization of the location of bacteria in relation to the epithelium (i.e., intracellular, adherent, or invasive).
There is an inherent bias in use of the 16S rRNA gene for the purpose of absolute quantification. The 16S rRNA genes of bacteria are organized into “operons” that vary in number from 1 to 15 among individual phylotypes. The operon number may also change during the growth phase and altered activity of cells.16,17 Consequently, molecular results should be related to absolute cell counts with caution. It is more appropriate to express quantitative results as relative proportions to either total bacteria or to other bacterial groups.
Metagenomics and Transcriptomics
Analysis of 16S rRNA genes has provided new information about the phylogenetic diversity of the intestinal microbiota. However, to understand the impact of the microbiota on GI health it is necessary to (a) identify members of the intestinal ecosystem, and (b) explore the functionality of the microbial community. Metagenomics and transcriptomics are emerging fields in microbiology that are based on high-throughput sequencing techniques or the use of microarrays (see Fig. 2-1). In metagenomics, DNA extracted from a sample is sequenced without prior amplification of specific genes. This results in a snapshot of the gene pool and functional potential of the microbiome. For example, metagenomic approaches have revealed the existence of a “core microbiome” in the intestine, because despite obvious differences in bacterial composition between individuals, these individuals share common microbial genes and metabolic pathways.18 In transcriptomics, mRNA is analyzed to provide a measure of gene expression within the intestinal microbiome. These techniques are expected to yield more in-depth understanding of microbial-host interactions in health and disease.
The Intestinal Ecosystem in Dogs and Cats
As a result of anatomical and physical differences, each intestinal compartment constitutes a unique ecosystem where microorganisms have their own niche and provide specialized functions by utilizing host nutrients and in return providing metabolites for host uptake (Table 2-1). Molecular studies reveal that each dog and cat has a unique microbial profile.6,19 The microbiota is similar at higher phylogenetic level between individual animals of the same species, but it differs substantially at the level of species and strain, with typically only 5% to 20% overlap in bacterial species between individual animals. Bacterial counts and diversity increase along the GI tract and may vary between the intestinal lumen and the mucosa.19,20 Bacterial counts vary between the fed and fasting state. The oral cavity is an important part of the intestinal ecosystem, because bacteria are constantly swallowed and they may be able to colonize parts of the intestine. The composition of the oral microbiota is complex. In one study, 84 different cultivable phylotypes were identified in the oral cavity of dogs, with the major groups being Actinomyces, Porphyromonas, Fusobacterium, Neisseria, and Streptococcus spp.21 Oral bacterial counts can reach up to 107 colony-forming units per gram (CFU/g). The stomach harbors 101 to 106 CFU/g, while bacterial counts in the duodenum and jejunum of dogs and cats range from 102 to 109 CFU/g. This is considerably higher than found in the human duodenum (<105 CFU/g). Cats appear to have higher counts of anaerobic bacteria in the small intestine compared with dogs.22 The ileum is a zone of transition between the small and large bowel and contains a more diverse microbiota and higher bacterial numbers (107 CFU/mL of contents) than the proximal small intestine. Colonic bacterial counts range between 109 and 1011 CFU/g of intestinal content. The predominant bacterial groups cultured from the canine and feline intestine include Bacteroides, Clostridium, Lactobacillus, Bifidobacterium spp., and Enterobacteriaceae (Table 2-2).
Table 2-1 Examples of Biochemical Reactions Performed by the Intestinal Microbiota
| Microbial Activity | Products | Representatives |
|---|---|---|
| Decarboxylation, deamination of amino acids | Ammonia | Clostridium spp., Peptostreptococcus spp., Peptococcus spp. |
| Deconjugation/dehydroxylation of bile acids | Secondary bile acids (cholate/deoxycholate) | Clostridium hiranonis, Lactobacillus spp. |
| Vitamin synthesis | Vitamins K2, B12, biotin, folate | Enterococcus spp., Pseudomonas spp., Sphingomonas spp., Lactobacillus spp. |
| Carbohydrate fermentation | Lactate, propionate, acetate, butyrate | Clostridium cluster XIVa, Prevotella spp., Faecalibacterium spp., Bifidobacterium spp. |
| Amino acid fermentation | Hydrogen, methane, amines, phenols, ammonia (NH3), organic acids, hydrogen sulfite | Sulfate-reducing bacteria (SRB), Desulfovibrio spp., Clostridium spp., Peptostreptococcus spp. |
| Degradation of oxalate | Formate and CO2 | Oxalobacter formigenes |
| Inulin and starch degradation | Lactate | Bifidobacterium spp. |
| Metabolism of alcohols and acetic acid | Methane and CO2 | Methanobacteria |
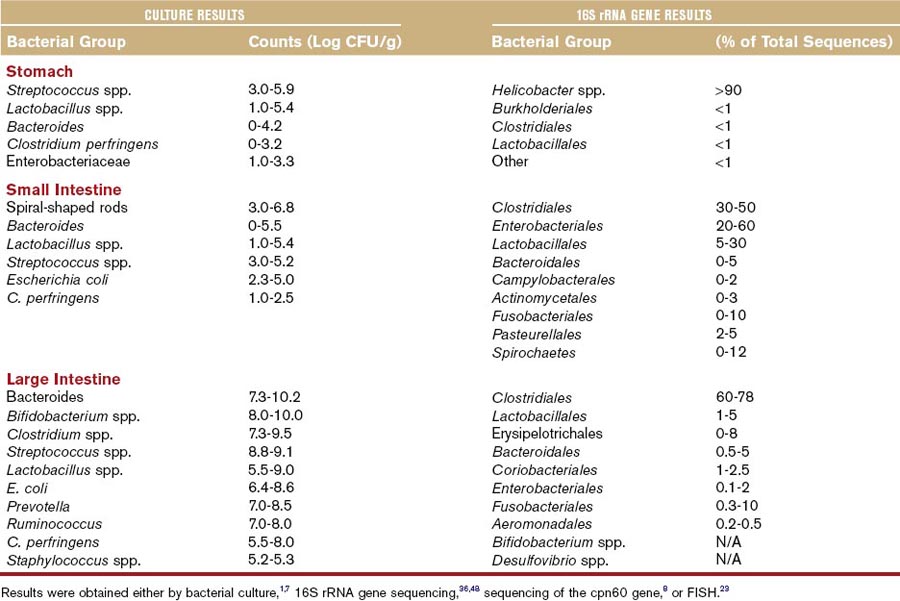
Because the vast majority of intestinal bacteria are not cultivable, molecular analysis (typically based on characterization of 16S rRNA) has expanded knowledge of diversity within the mammalian gut.2,7 Several thousand individual phylotypes are estimated to inhabit the human colon.1 There are approximately up to 900 bacterial phylotypes in the canine jejunum.2 Despite this vast diversity, only 12 of the 55 known major phylogenetic lineages have been observed in the mammalian GI tract (see Table 2-2). The phyla Firmicutes, Bacteroidetes, Proteobacteria, Actinobacteria, Spirochaetes, and Fusobacteria constitute almost 99% of all gut microbiota in dogs and cats. The remaining 1% is represented by the phyla Tenericutes, Verrucomicrobia, TM7, Cyanobacteria, Chloroflexi, and a few unclassified bacterial lineages. The relative proportions of these groups vary along the GI tract (see Table 2-2). Generally, proportions of aerobic bacteria or facultative anaerobic bacteria are higher in the proximal intestine, while anaerobes predominate in the colon. In the stomach, mucosa-adherent Helicobacter spp. predominate, followed by various lactic acid bacteria (e.g., Lactobacillus and Streptococcus spp.) and Clostridium spp. The proximal small intestine is more diverse than the stomach and harbors approximately 10 different bacterial phyla, with Clostridia, Lactobacillales, and Proteobacteria dominating.2 Proteobacteria and Spirochaetes are present in higher proportion in the proximal GI tract and typically represent <1% of sequences in the large intestine of healthy animals. Firmicutes is the major group represented in fecal samples (ranging between 30% to 95% of 16S rRNA gene sequences in various studies), followed by Bacteroides, Actinobacteria, and Fusobacteria. Firmicutes are a heterogenous bacterial phylum. They are represented mainly by the Clostridiales and Erysipelotrichaceae. Within those orders, Clostridium spp., Ruminococcus spp., Faecalibacterium spp., Dorea spp., and Turicibacter spp. are the major groups. Based on phylogenetic analysis, the Clostridiales are comprised of at least 70 different species (Table 2-3), which are organized into phylogenetically distinct Clostridium clusters. These clusters differ in abundance in different parts of the intestine. Clusters XIVa and IV encompass many important short-chain fatty acid-producing bacteria (see Table 2-3) and predominate in the ileum and colon of both cats and dogs. Cluster XI and I (Clostridium perfringens group) are the second most abundant groups in the small and large intestine of dogs and cats.6,7
Table 2-3 Most Abundant Representative of the Various Clostridium Clusters in Canine and Feline Fecal Samples
| Cluster I | Cluster XIVa |
| Clostridium perfringens | Dorea spp. |
| Clostridium colicanis | Roseburia/Ruminococcus group |
| Clostridium disporicum | Clostridium saccharolyticum |
| Clostridium celerecrescens | |
| Cluster IV | Clostridium symbiosum |
| Faecalibacterium spp. | Clostridium bolteae |
| Clostridium methylpentosum | Clostridium oroticum |
| Ruminococcus spp. | Clostridium methoxybenzovorans |
| Clostridium algidixylanolyticum | |
| Cluster XI | Clostridium hathewayi |
| Clostridium hiranonis | Clostridium amygdalinum |
| Clostridium bartlettii | Lachnospiraceae |
| Clostridium lituseburense | |
| Clostridium sordellii | Cluster XVIII |
| Clostridium glycolicum | Clostridium cocleatum |
| Peptostreptococcus spp. | Clostridium ramosum |
The order Clostridiales is the most abundant and most diverse group in the large intestine of dogs and cats, is comprised of at least 70 known species, and constitutes approximately one-third of total colonic bacteria.
Presence of a Core Microbiome and Functional Redundancy
There are marked differences in the composition of the microbiota between individuals, and even between monozygotic twins. However, the metabolic end products are similar between individuals. Additionally, although some environmental influences lead to significant changes in bacterial groups, these changes are not immediately associated with any obvious changes in gut function in healthy animals. New metagenomic studies have evaluated the gene content of the intestinal microbiota and suggest that the intestine harbors a “core microbiome,” because despite observed differences in bacterial phylotypes among individuals, the microbiome of each individual appears to have similar gene content and therefore similar functions.18 Furthermore, a functional redundancy exists in the GI tract. Several members of the community can perform similar functions, and if one group is displaced because of perturbations (e.g., antibiotic therapy), other members of the community are able to maintain a stable ecosystem. These findings highlight the need to evaluate the intestinal microbiome as an entity, including phylogenetic relationships and metabolic functions (i.e., metagenome, transcriptome, and metabolome).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


