Chapter 21 Fluid and Diuretic Therapy in Heart Failure
Advanced CHF, as well as the therapy of this syndrome, often is associated with alterations in renal function and a variety of fluid, electrolyte, and serum biochemical abnormalities. Some of these disturbances are mild and seemingly well tolerated, but others, such as hyponatremia and acute renal failure, indicate severe circulatory dysfunction and a need for urgent therapy.75 There are circumstances in which cardiac patients actually require fluid therapy to maintain optimal ventricular filling and prevent deterioration of renal function. However, it is more common for fluid therapy to produce edema or effusions in a previously compensated cardiac patient. Safe restoration of fluid and electrolyte balance in the patient with cardiovascular disease is challenging. To orchestrate such treatment, the clinician must appreciate the pathophysiology of heart failure and the compensatory changes that develop. This chapter addresses some of the clinically relevant pathophysiologic and therapeutic aspects of heart failure.
The normal circulation
The central circulation is regulated largely by a need to maintain plasma volume, mean ABP, and tissue perfusion. Of prime importance is the maintenance of normal effective plasma volume and ABP in the central circulation.141,150 These two variables depend on cardiac output, systemic vascular impedance, and renal regulation of sodium and water excretion. The reflexes that control the circulation have evolved so that blood pressure and plasma volume are maintained within a narrow range even in the presence of sudden physiologic stresses, such as exercise, hypotension, or hemorrhage. Blood pressure and plasma volume are monitored by different mechanoreceptors and osmoreceptors located in the arteries, veins, heart, kidney, and central nervous system. Ultimately, two factors—cardiac output and systemic vascular resistance (more precisely, vascular resistance and arterial impedance)—determine ABP (Figure 21-1). A change in either one of these two variables causes a parallel change in blood pressure. Numerous physiologic variables can affect cardiac output and vascular impedance (Box 21-1), and many of these factors are perturbed in CHF. Of particular relevance in this chapter are determinants of plasma volume in health and disease (Box 21-2). Plasma volume is a major contributor to venous pressure and cardiac filling. The serum sodium concentration, as described more fully in Chapter 3, plays a central role in determining plasma volume. Renal tubular activity, vascular dynamics, hormones, and other vasoactive factors regulate sodium balance. Abnormalities of sodium excretion are pivotal to the development of CHF.
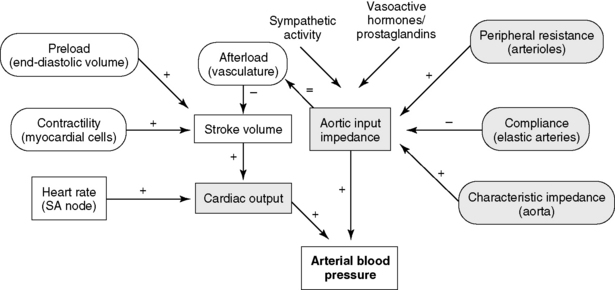
Box 21-1 Factors Controlling Arterial Blood Pressure
Box 21-2 Factors Regulating Plasma Volume in Heart Failure
Plasma Protein (albumin)Drug Therapy
Diuretics
Loop diuretics (furosemide, bumetanide, torsemide)
Thiazide diuretics (hydrochlorothiazide)
Potassium-sparing diuretics (triamterene, amiloride)
Spironolactone (blocks renal effects of aldosterone)
Carbonic anhydrase inhibitors (acetazolamide)
Angiotensin-converting enzyme inhibitors (captopril, enalapril, benazepril, lisinopril, ramipril)Digitalis glycosides, pimobendan, and other cardiotonic drugsVasodilator drugs (hydralazine, nitrates, angiotensin-converting enzyme inhibitors)
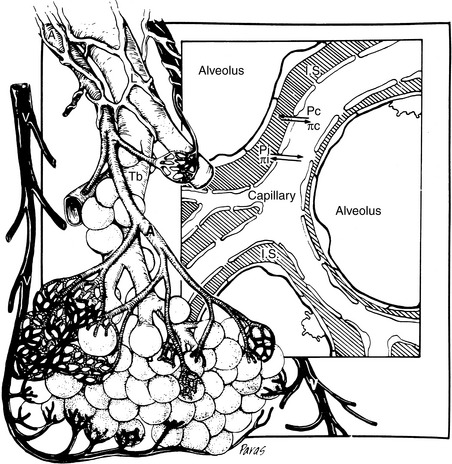
Attention also must be directed to the microcirculation and factors controlling fluid movement across capillaries. Tissue perfusion is crucial for organ functions such as the formation of urine, muscle contraction, and exchange of oxygen and carbon dioxide. Assuming the maintenance of adequate mean ABP, regional vascular resistance largely governs tissue perfusion across the arterial side of the microcirculation. Vascular resistance for any regional circulation is the sum of structural, autonomic, hormonal, and local vasoactive factors (see Box 21-1). Conversely, plasma volume and venous pressure exert the greatest effect at the venous end of the capillary. The interplay of hydrostatic pressures, oncotic pressures, capillary permeability, and lymphatic function determines whether the interstitium and serous body cavities accumulate or remain free of excess solute and water.75,156,165 The effect of these so-called Starling forces on fluid dynamics is summarized in Figure 21-2.
The circulation in heart failure
Heart failure is characterized clinically by hemodynamic abnormalities triggered by cardiac dysfunction.81 The causes of heart failure include numerous structural and functional disorders of the cardiac valves, myocardium, pericardium, and blood vessels, as well as sustained cardiac arrhythmias (Box 21-3). In response to impaired cardiac output, potent homeostatic mechanisms are activated that preserve perfusion of the brain and heart but at the expense of less vital regional circulations. Preservation of blood pressure mandates dramatic alterations in neural, hormonal, and cardiovascular function and structure. These adaptations (summarized in Box 21-4) include (1) activation of the sympathetic nervous system23,49,178 and release of hormones,46,121 (2) increased systemic vascular resistance47,48 and impedance,39 (3) reduction of autonomic reflex activity,81,186 and (4) cardiac dilatation and hypertrophy that together with myocardial interstitial changes are collectively referred to as “cardiac remodeling.54,76,113” Heart failure also alters renal function122,186 and enhances reabsorption of sodium and water.16,179 In combination, these potent control systems are capable of maintaining normal ABP in all but the most severe cases of cardiac failure.
Box 21-3 Causes of Heart Failure
Myocardial Diseases
Box 21-4 Neurohormonal, Renal, and Cardiovascular Activities in Congestive Heart Failure
Hormonal or Autocrine
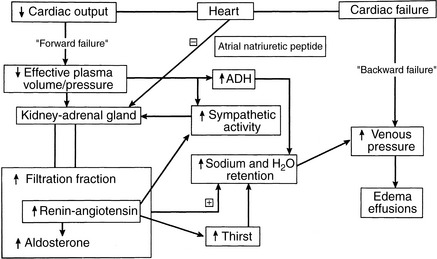
Hemodynamic abnormalities in the central circulation and microcirculation in CHF (Box 21-5) can be traced to both decreased cardiac performance and renal retention of sodium and water.122,141,152 Decreased cardiac output, valvular insufficiency, impaired myocardial relaxation, and reduced ventricular compliance increase ventricular end-diastolic pressure, which is transmitted back to the venous and capillary beds (“backward” failure). Higher venous and capillary pressures are augmented by renal fluid retention and expansion of the plasma volume. Renal sodium and water retention as a consequence of reduced cardiac output often is described in the medical literature as “forward” heart failure.141 Forward failure, in this regard, does not refer to clinical signs of low cardiac output but instead describes the renal responses triggered by low cardiac output. Forward failure is a critical factor in the development of edema and effusions in right-sided and biventricular heart failure. These concepts and some of the factors responsible for increased venous and capillary pressures are shown in Figure 21-3. The important role of the kidney in the pathogenesis of edema and effusions is discussed in the section on Renal Function in Heart Failure.
Box 21-5 Hemodynamic Consequences of Congestive Heart Failure
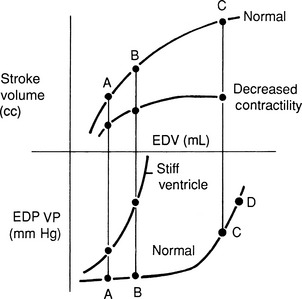
High venous pressures and increased ventricular end-diastolic pressure enhance cardiac filling and allow the ventricle to generate a greater contractile response. As shown in Figure 21-4, ventricular stroke volume is directly related to ventricular filling pressure.45,163 High venous pressure also maintains cardiac filling when ventricular compliance is decreased, as in hypertrophic cardiomyopathy, pericardial disease, or severe ventricular dilatation54 (see Figure 21-4, bottom). The clinical relevance of this relationship becomes obvious when the edematous patient is treated with diuretics and venous pressures, ventricular filling, and cardiac output decline, causing systemic hypotension or prerenal azotemia.
The term congestive heart failure implies a situation of increased venous and capillary hydrostatic pressures, increased transudation of fluid across capillary walls, and net accumulation of fluid in the interstitial compartment (i.e., edema) or serous body cavities (i.e., effusion). A safety margin normally prevents this accumulation of fluid, and venous pressures must increase substantially (usually to two or three times above the normal upper limit) before edema develops.60,157,165,180 Development of pulmonary edema in the dog usually requires left atrial pressure to increase acutely to more than 20 mm Hg.60 Substantial increases in lymphatic drainage permit much higher pressure to be tolerated chronically.16,34 In addition to increased venous pressures, hypoalbuminemia can contribute to edema formation.60,183 As a consequence of variable lymphatic drainage and other factors, such as capillary permeability and compartment compliance, edema is not uniformly distributed in the tissues.180 This nonuniform distribution is evident clinically inasmuch as acute cardiogenic pulmonary edema in the dog is most prominent in perihilar and in lung lobes on the rightside, although it can accumulate in cranial and ventral regions at the same time.
The edema of CHF develops predominantly in the capillary beds drained by the failing side of the heart. This finding is pertinent because CHF is classified clinically as left-sided, right-sided, or biventricular. Increased pulmonary venous and capillary hydrostatic pressures cause pulmonary edema (see Figure 21-2), the cardinal finding of left-sided CHF. Right-sided heart failure increases systemic venous pressures leading to jugular venous distention or pulsation, hepatic congestion, ascites, or (infrequently in small animals) subcutaneous edema. Increased systemic venous pressure even may contribute to pulmonary edema formation.103
Pleural effusions develop as a result of left-sided, right-sided, or, most often, biventricular failure. This finding can be explained by the dual venous drainage of the pleural surfaces (i.e., parietal drainage is systemic, whereas visceral drainage is pulmonary). Although veterinary textbooks usually attribute pleural effusion to isolated right-sided CHF, this is not common in human patients. Pleural effusion correlates better with pulmonary capillary wedge pressure than with right atrial pressure.183 Similarly, pleural effusions in small animals most often indicate biventricular CHF. Although pleural effusion does occur in some dogs and cats with predominantly right-sided cardiac disease (e.g., pulmonic stenosis, tricuspid malformation), ascites is more common in dogs. Clinically significant pleural effusions are rare in animals with isolated right ventricular failure caused by heartworm-induced pulmonary hypertension.15,169 Conversely, pleural effusions are common when end-stage CHF develops in dogs with severe mitral regurgitation, pulmonary hypertension, and secondary right ventricular dysfunction or in cats with any form of severe cardiomyopathy. Pleural effusion may become chylous in nature in those with advanced CHF.
In contrast to right-sided or biventricular CHF, severe left-sided heart failure can develop without substantial sodium retention or weight gain.69 Two common examples in veterinary medicine can be cited. The first example is rupture of a mitral chorda tendinea in an older dog with previously stable mitral regurgitation. The sudden increase in mitral regurgitant volume increases mean left atrial and pulmonary capillary pressures, leading to peracute pulmonary edema. The second example is a cat with hypertrophic cardiomyopathy and a noncompliant left ventricle (see Figure 21-4, bottom left curve). It is not uncommon for severe pulmonary edema to follow a bout of protracted tachycardia (e.g., stress). Development of pulmonary edema in these situations can be explained by acute deterioration of left ventricular systolic or diastolic performance that rapidly increases left atrial and pulmonary venous pressure. Although diuresis is a critical treatment in this situation, short-term success may hinge on therapy that reduces mitral regurgitant fraction (i.e., afterload reduction).
Another issue of relevance to CHF and fluid therapy of the cardiac patient is the relative size of the vascular compartments. The vascular compliance of the pulmonary circulation is much smaller than that of the systemic circulation, and sudden expansion of the plasma volume usually increases pulmonary venous pressure more than systemic venous pressure. This is particularly true in the patient with left-sided heart disease and explains why some dogs and cats develop pulmonary edema after intravenous administration of a so-called maintenance volume of crystalloid solution. Furthermore, central venous pressure (CVP) cannot be used to gauge the effect of intravenous fluid therapy on left-sided cardiac filling pressures, especially in the setting of isolated left-sided CHF.141 Owing to differences in vascular compliance and cardiac function, left-sided filling pressures may increase much more rapidly than CVP, though both increase simultaneously.
Renal function in heart failure
Remarkably, the kidney often is able to maintain glomerular filtration in the setting of decreased blood pressure or cardiac output. Decreases in renal perfusion are countered by dilatation of the afferent arteriole mediated by the release of prostaglandin E2, and constriction of the efferent arteriole primarily by angiotensin II. Efferent arteriolar constriction also is augmented by arginine vasopressin (antidiuretic hormone [ADH]) and norepinephrine.122 These microvascular responses increase glomerular filtration pressure, increase filtration fraction, and maintain glomerular filtration in the setting of reduced renal blood flow (see Chapter 2).116,138,159 However, considering normal renal function demands approximately 20% of a normal cardiac output, it is not surprising to identify azotemia in advanced CHF, especially during aggressive diuretic therapy. Progressive renal failure is common in dogs and cats with CHF, and the treatment of patients with both intrinsic renal disease and heart failure is especially difficult. This situation also occurs in human patients in whom worsening of renal function is associated with a poorer prognosis and higher mortality and often requires dialysis for control of both problems.57 Neurohormones and cytokines, angiotensin in particular, are considered central to the progression of renal disease in heart failure.80 Aggressive therapy of heart failure may slow progression of renal disease in humans.89
The renal response to decreased cardiac output is central to the pathogenesis of edema and effusions in heart failure. Studies of induced right-sided heart failure and spontaneous CHF in dogs have demonstrated avid retention of administered salt loads.9,84 Numerous mechanisms have been identified for persistent sodium retention in CHF (see Figure 21-3). These alterations include redistribution of renal blood flow,9,131 enhanced tubular sodium reabsorption,8,91,102 release of prostaglandins,36,38,111 greater renal sympathetic nerve activity,* increased renal interstitial pressure,58,101 and increased hormonal activity. The last includes increases in vasopressin (ADH),16,136 angiotensin II, and aldosterone† (see Box 21–4). Presumably, these mechanisms also operate in animals with spontaneous heart failure.134
Particular emphasis has been placed on the increased concentrations of renin, angiotensin II, and aldosterone found in patients with CHF.121 There are a number of triggers for the release of renin in the cardiac patient.113 One mechanism is the stimulation of renal ß-adrenergic receptors by sympathetic efferent traffic activated in response to hypotension. Renin also is released in response to reduced renal blood flow related to heart failure or volume depletion caused by diuretic therapy of CHF.182 Severe sodium restriction, especially in dogs with signs of heart disease but without overt CHF, can lead to renin release.127 Clinically, the effects of angiotensin II and aldosterone can be mitigated in part by drugs that inhibit formation of angiotensin II (angiotensin-converting enzyme [ACE] inhibitors such as enalapril and benazepril) or drugs that block the AT-1 receptor of angiotensin II, such as losartan and candesartan.
Other factors promote renal fluid retention in CHF. Changes in intrarenal blood flow can lead to redistribution of flow to the salt-conserving juxtamedullary nephrons.16,91,93 Increased filtration fraction maintains the glomerular filtration rate (GFR) but predisposes to renal tubular reabsorption of water (see Chapter 2). Arginine vasopressin (ADH) also plays a role. In CHF, increases in plasma ADH concentration probably represent nonosmotic release in response to low ABP.122 Increased thirst (mediated by angiotensin II), when combined with increases in ADH, can contribute to free-water retention and hyponatremia.35,99,120 Endothelin is another hormone released from endothelial cells in CHF.132,176 This hormone reduces renal blood flow, the GFR, and urinary sodium excretion.98,122 The sequence in which these mechanisms are activated varies with the type and severity of heart failure.46,141 However, it is clear that with deterioration in cardiac function, sodium- and water-retaining mechanisms are exacerbated, and further expansion of the plasma volume occurs. Blunting the renal response generally requires appropriate medical treatment of CHF, progressive restriction of dietary sodium, and administration of diuretics.
In CHF, the vasoconstrictive and sodium-retaining mechanisms overwhelm local and systemic vasodilator and natriuretic systems. Distention of the atria and ventricles signals release of atrial natriuretic peptide (ANP) and brain natriuretic peptide (BNP). These peptides of cardiac origin stimulate formation of cyclic GMP, leading to diuresis, vasodilatation, and improved ventricular relaxation.107,113 Although increased circulating concentrations of ANP and BNP can be measured in dogs with experimentally induced and spontaneous CHF,* it also has been shown that the renal response to these hormones is blunted or antagonized. 21,104,135 If dogs or people with CHF are treated with pharmacologic doses of ANP, however, or if the degradation of ANP is reduced by administration of a neutral endopeptidase inhibitor, diuresis may follow.104,110 Other vascular-modulating factors, such as the vasodilator nitric oxide, are more difficult to assess in CHF, but metabolites of this endothelial-derived substance reportedly are decreased in some dogs with mitral regurgitation.126
Cardiovascular drugs and renal function
Effects of diuretics on renal function
Diuretics used in management of CHF prevent reabsorption of solute and water, leading to increased urine flow. Diuretics are essential to both the short- and long-term management of CHF. While there are no blinded clinical trials evaluating the efficacy of furosemide, experienced clinicians repeatedly observe the short-term benefit of furosemide diuresis in veterinary patients with life-threatening pulmonary edema. In a 3-week study of dogs with CHF, pulmonary wedge pressure, an estimate of left atrial pressure, declined within hours after initiating furosemide and remained below baseline after 21 days of therapy.71 Despite the lack of study in terms of chronic efficacy, diuretics are a mainstay for acute and chronic treatment of CHF in dogs, cats, and humans.70 They reduce preload by decreasing cardiac filling (venous) pressures and chronically prevent the excessive dietary sodium retention characteristic of CHF,9 allowing for a new steady state of sodium balance to develop.70 Diuretics may also reduce left ventricular afterload by reducing sodium loading and vascular resistance in arterioles.
The clinical pharmacology of these drugs and effects on renal function (Tables 21-1 and 21-2) are relevant to understanding their effectiveness and limitations. All of the commonly used diuretics, except spironolactone, are delivered by renal blood flow and secreted as organic acids into the proximal tubule. Circulatory failure, reduced renal blood flow, administration of nonsteroidal antiinflammatory drugs (NSAIDs), or primary renal failure may reduce the renal delivery of a diuretic. In the case of renal failure, endogenous organic acids can compete with furosemide for transport across the proximal nephron. Once secreted into the filtrate, a diuretic inhibits salt and water transport via a specific mechanism and at relatively specific sites along the nephron.72,112,142
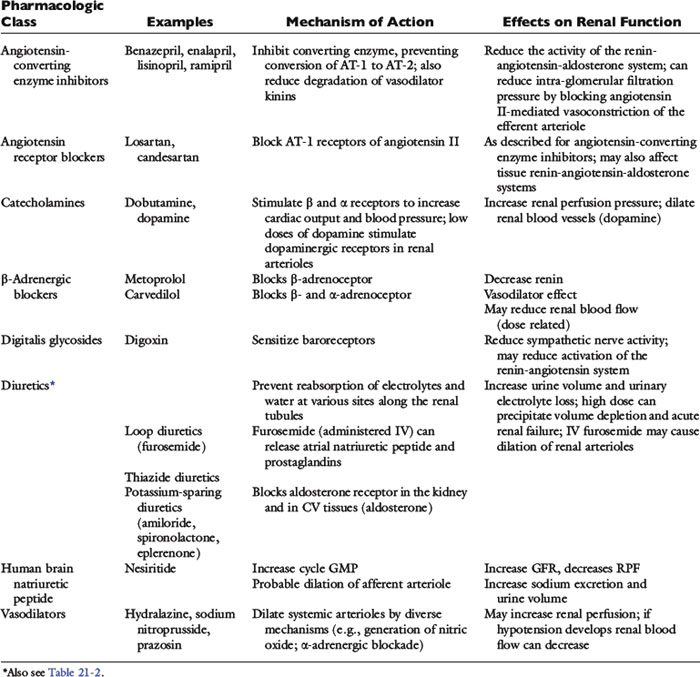
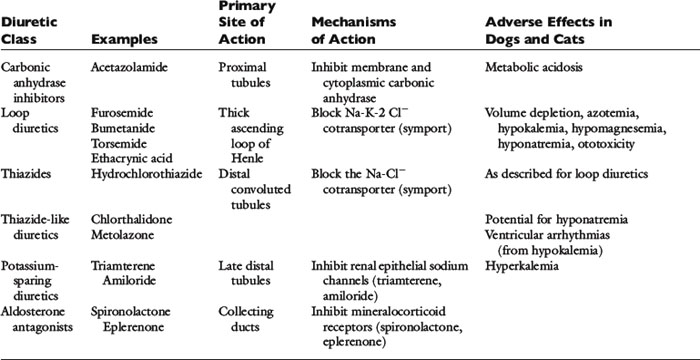
Figure 21-5 demonstrates the general sites of action of the commonly used diuretics. The importance of understanding these details can be illustrated by two examples. First, the effectiveness of a diuretic depends on the ability of cells distal to the site of diuretic action to reabsorb sodium and water. Initially in CHF, loop diuretics, which act on the thick portion of Henle’s loop, are highly effective. However, in severe chronic CHF, the more distal tubular cells can increase their reabsorption of sodium and water and overcome the effects of the diuretic.141 This problem can be counteracted with additional treatment such as the combination of hydrochlorothiazide and spironolactone, which act more in the distal nephron. This type of sequential nephron blockade can induce a marked diuresis in some dogs but not without risk of volume depletion and acute renal failure. A second example pertains to the adverse effects of diuretics. Loop diuretics increase the delivery of sodium to cells of the late distal convoluted tubules and collecting ducts. At those sites, sodium is reabsorbed in exchange for potassium (under the influence of aldosterone) or hydrogen ions that are secreted.72 These ion exchange mechanisms have the potential to cause hypokalemia and metabolic alkalosis, especially with high doses or chronic therapy.
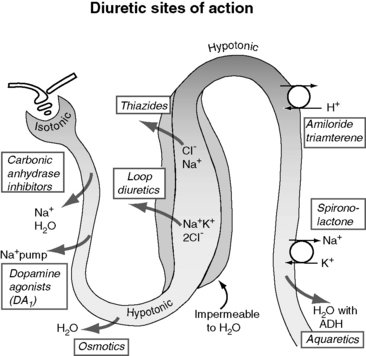
Furosemide, ethacrynic acid, bumetanide, and torsemide exert their effects on the ascending limb of Henle’s loop.112,142,170 These so-called loop diuretics block the Na+-K+-2 Cl− cotransporter (symport) and prevent the active transport across the tubular lumen of two chloride ions, one sodium ion, and one potassium ion. Loop diuretics are potent with a good dose response, whereas higher dosages result in greater sodium loss (“high ceiling”). This is related to the high capacity for reabsorbing filtrate at this site (normally approximately 25% of the filtrate is reabsorbed there). Urinary concentration is impaired because blocking the Na+-K+-2 Cl− carrier impedes development of a hypertonic renal interstitium. Urinary dilution also is impaired because dilution of the filtrate is a normal function of this segment. After administration of a loop diuretic, there are substantial losses of chloride, sodium, water, and other electrolytes (including potassium, magnesium, and calcium) in the urine. In addition to tubular effects, some hemodynamic and extrarenal effects of loop diuretics may arise from vasodilatation or increased venous capacitance.32,70 For example, intravenous administration of furosemide releases vasodilator prostaglandins that increase venous capacity and renal blood flow.13,109,117,124 Dyspnea in human patients is relieved even before a reduction in lung water can be identified.70
The thiazide and thiazide-like diuretics act on the cortical distal convoluted tubule by competing with the luminal Na-Cl cotransporter and preventing movement of NaCl into the distal tubular cells. This effect impairs the ability to dilute urine (and excrete solute-free water) but does not necessarily affect urine concentration, which is a medullary function. Accordingly, in hyponatremia, when there is impaired free-water clearance, the thiazide diuretics are relatively contraindicated.75,142 The overall potency of thiazide diuretics is limited, in part because about 90% of the filtrate already has been reabsorbed before the distal nephron has been reached. For this reason these agents are low ceiling agents, and in human patients higher dosages do not lead to proportionate losses in sodium (but do predispose to more serious losses of potassium and magnesium).70 Nevertheless, when even low doses of a thiazide diuretic is combined with furosemide, dangerous volume depletion and electrolyte losses can result.
The late distal tubules and collecting ducts are sites of sodium reabsorption, aldosterone-controlled secretion of potassium ions, ADH-mediated water reabsorption, and urine concentration. Diuretics acting at these sites initiate diuresis by preventing movement of sodium through luminal channels, either by directly blocking the channels (e.g., amiloride, triamterene) or by antagonizing the effect of aldosterone (e.g., spironolactone, eplerenone). These drugs also exert a potassium-sparing effect and often are classified by that description. They act very distally in the nephron, and their quantitative potential to inhibit sodium reabsorption is low, resulting in a weak to nearly absent diuretic effect.74 The diuretic effect of spironolactone depends on prevailing aldosterone concentrations (which are low in animals with mild CHF or in those receiving appropriate dosages of ACE inhibitors). The main value of these drugs is for maintenance of normal serum potassium concentration or antagonism of aldosterone-induced cardiac injury.129,160,161,181
There are a number of clinically relevant issues regarding the dosage and administration of diuretics.30,142,153,158 Many of the commonly used diuretics are organic anions at physiologic pH and are highly bound to serum proteins. To be effective, the diuretic must be delivered to the urinary space by glomerular filtration or active secretion in the proximal renal tubule. Active secretion is the more important mechanism because the drug is concentrated in tubular fluid. Reduced renal perfusion associated with heart failure, as well as primary renal disease or NSAID administration, may limit the effectiveness of a diuretic unless a high dosage is used and the drug is sufficiently concentrated in renal tubular fluid. This concern about renal perfusion is one rationale for initial high-dose, intravenous administration of furosemide in patients with life-threatening CHF. It also explains in part why chronic diuresis may be associated with impaired response to diuretics. Drug delivery is also relevant in terms of oral dosing of diuretics. Gastrointestinal absorption may be impaired in CHF, especially with right-sided failure and intestinal edema. More importantly the time course from absorption to renal delivery is longer than for parenteral administration. This reduces the concentration of the drug acting at the tubules, decreasing the overall diuretic effect. Temporarily switching from oral to parenteral administration of furosemide at the same dose can have a dramatic diuretic benefit in some patients because of the more efficient delivery of the drug. Additional clinical situations in which diuretics may fail include treatment of pain with opiates (which stimulate ADH release), unanticipated high sodium intake, and acute worsening of heart failure. In these situations the diuretic dosage required to establish diuresis successfully may be substantially higher or an alternative route of administration may be required.
Diuretic therapy triggers neurohormonal responses,46,149,182 and diuretic monotherapy is not an appropriate management strategy for long-term treatment of CHF. Diuretic-induced volume depletion invariably leads to a rebound in renal retention of salt and water either at the previous or a new steady-state in terms of sodium balance. This concept, termed the braking phenomenon, is highly important for understanding the basis for multidrug therapy and why furosemide is typically given two or even three times daily. As an example, once-daily dosing of furosemide in human patients is associated with a brisk diuresis for about 6 hours. But over 24 hours there may be no net loss in total body sodium or edema because salt and water retention can occur for the balance of the day.70 This effect is mediated partly by decreased tubular flow rate, salt retention in segments of the nephron unaffected by the diuretic used, increased sympathetic activity, and activation of the renin-angiotensin-aldosterone system (RAAS).112,141 Thus control of edema in CHF requires a steady state of reduced sodium retention, and patients should receive a consistent dosage of furosemide along with an ACE inhibitor, spironolactone, and a sodium restricted diet.
The dosage of diuretics used must be effective but should be carefully controlled to minimize the common complications of dehydration, azotemia, electrolyte imbalance, and potentially deafness. The first dosage of furosemide chosen for a patient with life-threatening pulmonary edema often is high (2 to 5 mg/kg, intravenously every 1 to 3 hours) to ensure diuresis. The furosemide dosage is promptly reduced if symptomatic improvement and a brisk diuresis are observed. These effects can occur within 1 to 2 hours of administration of furosemide,71 but a lag period (12 to 24 hours) may be noted between obvious clinical improvement and clearing of radiographic pulmonary densities. Owing to the potential for overzealous diuresis and iatrogenic renal failure and electrolyte disturbances, the clinician should evaluate serum biochemistries every 24 to 48 hours until the patient is eating and drinking satisfactorily. After a stable diuretic course of 2 weeks, most dogs and cats maintain relatively stable renal function and serum potassium concentrations unless a decompensating factor (e.g., vomiting, anorexia) intervenes. This is especially true when ACE inhibitors and spironolactone are prescribed concurrently because they reduce aldosterone concentration or effect and decrease potassium and magnesium losses. Thus stable serum creatinine and potassium concentrations over two or three reevaluation periods are likely to be maintained for some time.141 The overall dosage of diuretics in dogs should be limited by using combination therapy for CHF, including progressive sodium restriction, ACE inhibitors, spironolactone, and pimobendan.10,61,62,78,95,123 Cats with chronic CHF typically receive furosemide, an ACE inhibitor, and sometimes pimobendan or spironolactone. Cats receiving furosemide are more prone to develop mild to moderate azotemia and hypokalemia than are dogs, even at dosages that are 50% lower than daily dosages typically used for dogs. Spironolactone is usually well tolerated in cats but may cause anorexia or ulcerative skin lesions.97
Effects of other cardiovascular drugs on renal function
Normal autonomic responses to changes in blood pressure and normal heart rate variability are blunted in CHF.66,94 This is associated with dominant sympathetic activity in cardiac failure. Sympathetic nerve activity can increase renin release and affect renal blood flow.122 Digitalis glycosides such as digoxin appear to exert a neurotropic effect and restore baroreceptor sensitivity and parasympathetic tone, and this effect is independent of the inotropic action of the drug.81,166 By this or some other effect, digoxin also can blunt the RAAS in CHF. Although digoxin has been largely supplanted by the inodilator, pimobendan, due to this autonomic effect, digoxin therapy maintains a role in patients with atrial fibrillation and end-stage heart failure.
Cardiac patients sometimes are treated with aspirin and other antiprostaglandin drugs to prevent blood clots (cats) or to alleviate signs of osteoarthritis (dogs). These NSAIDs may be deleterious when used in CHF patients. By preventing prostaglandin-induced dilatation of the afferent arteriole, NSAIDs may decrease glomerular filtration pressure. They may be especially hazardous when used in combination with furosemide and ACE inhibitors. Practically speaking, the use of cyclooxygenase (COX) inhibitors can be tolerated in heart failure, but it is prudent to start at one third to one half of the normal dose and increase the dose while monitoring renal function (and for signs of gastrointestinal ulceration). Other drugs, including ß-adrenergic blockers, human BNP, neutral endopeptidase inhibitors, and direct vasodilators, also may affect renal function. Some of the major effects of these drugs are summarized in Table 21-1. The effects of BNP on renal hemodynamics are still under investigation, and these effects may differ in normal subjects from those in patients with CHF or systemic hypertension.73,172
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


