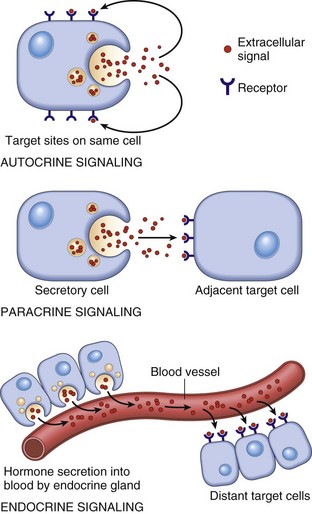
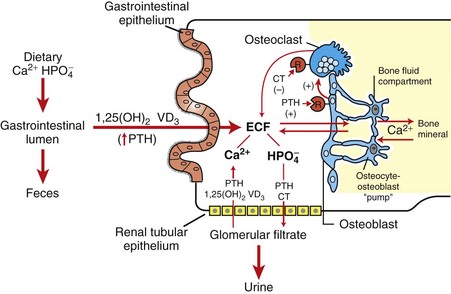
CHAPTER 12 Signaling molecules are grouped into three general categories according to the source of the signal and the location of the target on which the signal has an effect (Fig. 12-1). In autocrine signaling, cells respond to signals that they themselves secrete. Molecules produced by one cell that act on a neighboring cell are characteristic of paracrine signaling. The final pattern of signaling, which is the focus of this chapter, is endocrine signaling, whereby hormones produced by cells of endocrine organs are released into the circulation and act on distant target cells. The adenohypophysis consists of three portions: the pars distalis, pars tuberalis, and pars intermedia (Fig. 12-2). In many animal species, the adenohypophysis completely surrounds the pars nervosa of the neurohypophyseal system. The pars distalis is the largest and is composed of several different endocrine cell populations surrounded by abundant capillaries to facilitate secretion of their trophic hormones (Web Fig. 12-1). Fig. 12-2 Pituitary gland and brainstem, normal dog. Web Fig. 12-1 Pituitary gland, pars distalis, normal dog. A specific population of endocrine cells is present in the pars distalis (and also in the pars intermedia of dogs for ACTH secretion) that synthesizes, processes, and secretes each of the pituitary trophic hormones (Fig. 12-3). Secretory cells in the adenohypophysis are classified as acidophils, basophils, and chromophobes based on the reactions of their secretory granules with pH-dependent histochemical stains (Fig. 12-4). Based on contemporary specific immunohistochemical staining, acidophils can be further subclassified functionally into somatotrophs that secrete growth hormone (GH; somatotrophin) and lactotrophs that secrete prolactin. Basophils include gonadotrophs that secrete luteinizing hormone (LH) and follicle-stimulating hormone (FSH), thyrotrophs that secrete thyrotrophic hormone (thyroid-stimulating hormone [TSH]), and ACTH-secreting corticotrophs. Chromophobes are pituitary cells that by light microscopy lack stainable cytoplasmic secretory granules. They include the pituitary cells involved with the synthesis of ACTH and melanocyte-stimulating hormone (MSH) in some species, nonsecretory follicular (stellate) cells, degranulated chromophils (acidophils and basophils) in the actively synthesizing phase of the secretory cycle, and undifferentiated stem cells of the adenohypophysis. Fig. 12-3 Hypothalamic-pituitary-target gland axis. Fig. 12-4 Pars distalis, normal dog. Each type of endocrine cell in the adenohypophysis is under the control of a specific releasing hormone or factor from the hypothalamus (see Fig. 12-3). These releasing hormones are small peptides synthesized and secreted by neurons of the hypothalamus. They are transported by axonal processes to the median eminence, where they are released into capillaries and conveyed by the hypophyseal portal system to specific endocrine cells in the adenohypophysis. Each hormone stimulates the rapid release of secretory granules containing a specific preformed trophic hormone. The adrenal glands of mammals consist of two distinct parts that differ not only in morphology and function but also in embryologic origin. Because of their close structural relationships, the outer cortex and inner medulla of the adrenal glands usually have been considered parts of one organ (Fig. 12-5). The adrenal cortex develops from cells of the coelomic epithelium that are of mesodermal origin. The chromaffin tissue and sympathetic ganglion cells of the adrenal medulla are derived from ectoderm of the neural crest. Fig. 12-5 Adrenal gland, normal dog. Mineralocorticoids are adrenal steroids that principally affect ion transport by epithelial cells and cause excretion of potassium and conservation of sodium. The most potent and important naturally occurring mineralocorticoid is aldosterone. Enzymatically controlled electrolyte pumps in epithelial cells of the renal tubule and sweat glands respond to mineralocorticoids by conserving sodium and chloride and by excreting potassium. In the distal convoluted tubule of the mammalian nephron, a cation-exchange mechanism is responsible for the resorption of sodium from the glomerular filtrate and secretion of potassium into the lumen (Fig. 12-6). These reactions are accelerated by mineralocorticoids but still proceed, although at a much slower rate in their absence. Lack of mineralocorticoid secretion, such as in the Addison’s-like disease of dogs, can result in lethal retention of potassium and loss of sodium. Fig. 12-6 Aldosterone secreted by the zona glomerulosa of the adrenal cortex acts on the distal portions of the nephron to increase tubular excretion of potassium and increase resorption of sodium (and secondarily of chloride). Glucocorticoids exert a profound negative effect on wound healing. Dogs with hypercortisolism can have wound dehiscence (Fig. 12-7). The basic mechanism is inhibition of fibroblast proliferation and collagen synthesis, leading to a decrease in scar tissue formation. Fig. 12-7 Dehiscence of surgical wound, skin, ventral abdomen, dog. The thyroid gland is the largest of the endocrine organs that function exclusively as endocrine glands. The basic histologic structure of the thyroid gland is unique among endocrine glands and consists of follicles of varying size (20 to 250 µm), which contain colloid produced by the follicular cells. The follicular cells are cuboidal to columnar and are orientated so that their secretory pole is directed toward the lumen of the follicle. An extensive network of interfollicular and intrafollicular capillaries provides the follicular cells with an abundant blood supply. Follicular cells have extensive profiles of rough endoplasmic reticulum for synthesis and a large Golgi apparatus for packaging of substantial amounts of protein, which are then transported into the follicular lumen. The luminal side of follicular cells in contact with the colloid has numerous microvilli (Web Fig. 12-2). Web Fig. 12-2 Thyroid follicular cells, thyroid gland, normal dog. The synthesis of thyroid hormones is unique among those of the endocrine glands because the final assembly of hormone occurs extracellularly within the follicular lumen. Follicular cells trap essential raw materials, such as iodide from the blood, by a sodium-iodide symporter in the basolateral plasma membrane and then transport them rapidly against a concentration gradient to the lumen, where the iodide is oxidized by thyroid peroxidase in the microvilli to iodine (I2) (Fig. 12-8). The assembly of thyroid hormones within the follicular lumen is made possible by a unique protein, thyroglobulin. Thyroglobulin is a high molecular weight (600,000 to 750,000 Da) glycoprotein synthesized in successive subunits on the ribosomes of the endoplasmic reticulum in follicular cells. The constituent amino acids (tyrosine and others) and carbohydrates (e.g., mannose, fructose, galactose) are derived from the circulation. Recently synthesized thyroglobulin (17S) leaves the Golgi apparatus and is packaged into apical vesicles that are extruded into the follicular lumen (see Fig. 12-8). The amino acid tyrosine, an essential component of thyroid hormones, is incorporated within the molecular structure of thyroglobulin. Iodine is bound to tyrosyl residues in thyroglobulin at the apical surface of follicular cells to form monoiodotyrosine (MIT) and diiodotyrosine (DIT) (see Fig. 12-8). The resulting MIT and DIT combine to form the two biologically active iodothyronines, T4 and T3, secreted by the thyroid gland. Fig. 12-8 Thyroid follicular cells illustrating two-way traffic of materials from capillaries into the follicular lumen. TSH is delivered to thyroid follicular cells where it binds to the basilar aspect of the cell, activates adenyl cyclase, and increases the rate of all biochemical reactions concerned with the biosynthesis and secretion of thyroidal hormones. If the secretion of TSH is sustained (hours or days), thyroid follicular cells become more columnar and follicular lumina become smaller as a result of increased uptake of colloid by endocytosis (Fig. 12-9). Numerous periodic acid–Schiff (PAS)-positive colloid droplets are present in the luminal aspect of the hypertrophied follicular cells. The converse occurs in the thyroid gland in response to increases in circulating T4 and T3, which cause a corresponding decrease in TSH. Thyroid follicles become enlarged and distended with colloid as a result of decreased TSH-mediated endocytosis of colloid. Follicular cells lining the involuted follicles become low cuboidal, with only a few endocytic vacuoles at the interface between the colloid and follicular cells (Fig. 12-10). Fig. 12-9 Hyperplasia, thyroid gland, horse. Fig. 12-10 Atrophy, thyroid gland, dog. Calcitonin is secreted by a second endocrine cell population, C or parafollicular cells, in the mammalian thyroid gland. These cells are situated either in the follicular wall, within the basement membrane between follicular cells, or in small groups adjacent to interfollicular capillaries between follicles (Web Fig. 12-3). C cells do not border the follicular colloid directly, and their secretory pole is oriented toward the interfollicular capillaries. The distinctive feature of C cells is the presence of numerous small, membrane-limited, cytoplasmic secretory granules, which are immunoreactive for calcitonin. Web Fig. 12-3 Thyroid C cell, thyroid gland, normal dog. C cells store substantial amounts of calcitonin in their cytoplasm, and the hormone is discharged rapidly into interfollicular capillaries in response to hypercalcemia (Fig. 12-11). C cells respond to long-term hypercalcemia by hyperplasia. When the blood calcium concentration is reduced, the stimulus for calcitonin secretion is diminished, and numerous secretory granules accumulate in the cytoplasm of C cells (see Fig. 12-11). Calcitonin exerts its function by interacting with target cells located primarily in bone and kidneys. The actions of parathyroid hormone (PTH) and calcitonin are antagonistic on bone resorption, but synergistic in decreasing the renal tubular reabsorption of phosphorus. Fig. 12-11 Response of thyroid C cells and parathyroid chief cells to hypercalcemia and hypocalcemia. The parathyroid glands of animals are composed predominantly of chief cells in different stages of secretory activity (Fig. 12-12). Oxyphil cells, often forming nodules, are also present in parathyroid glands of senile horses and cattle. They are larger than chief cells, and their abundant cytoplasm is filled with numerous large, often bizarre-shaped mitochondria and few secretory granules. Although their presence has been associated with hyperparathyroidism in certain species, the extent of their functional capacity remains controversial. Fig. 12-12 Parathyroid gland, normal dog. Biologically active PTH secreted by chief cells is a straight-chain polypeptide consisting of 84 amino acid residues, with a molecular weight of approximately 9500 Da. Secretory cells in the parathyroid glands of most animals store relatively small amounts of preformed hormone but are capable of responding to minor fluctuations in calcium ion concentration rapidly, by altering the rate of hormonal secretion, and more slowly, by altering the rate of hormonal synthesis (Fig. 12-13). In contrast to most endocrine organs that are under complex control, the parathyroid glands have a unique feedback control system based primarily on the concentration of calcium and, to a lesser extent, of magnesium ions in blood. Calcium ion concentration controls not only the rate of biosynthesis and secretion of PTH but also other metabolic and intracellular degradative processes within chief cells. Increased calcium ion concentration in extracellular fluids rapidly inhibits the uptake of amino acids by chief cells, and consequently synthesis of proPTH, its conversion to PTH, and secretion of stored PTH (see Fig. 12-13). Fig. 12-13 Bypass secretion of parathyroid hormone (PTH) in response to increased demand signaled by decreased blood calcium ion concentration. PTH is the principal hormone involved in the minute-to-minute, fine regulation of blood calcium concentration (total and ionic calcium) in mammals. It does this by directly influencing the function of target cells located primarily in bone and the kidneys, and indirectly acting in the intestine to maintain plasma calcium concentration sufficient to ensure the optimal functioning of a wide variety of body cells. The overall action of PTH on bone is to mobilize calcium into extracellular fluids (Fig. 12-14). Bone responds to PTH by increasing the activity of osteoclasts and osteocytes existing in bone. The third major hormone involved in the regulation of calcium metabolism and skeletal remodeling is cholecalciferol or vitamin D3 (see Fig. 12-14). Cholecalciferol is ingested in small amounts in the diet and can be synthesized in the epidermis from precursor molecules (e.g., 7-dehydrocholesterol) through a provitamin D3 intermediate form in response to ultraviolet light. The active metabolites of vitamin D increase the absorption of calcium and phosphorus from the intestine and thereby maintain adequate concentrations of these electrolytes in the extracellular fluids as required for the appropriate mineralization of bone matrix. From a functional point of view, vitamin D brings about the retention of sufficient mineral ions to ensure mineralization of bone matrix, whereas PTH maintains the proper ratio of calcium to phosphorus in extracellular fluids. The major target tissue for 1,25-(OH)2D3 is the mucosa of the small intestine, where it increases the active transcellular transport of calcium (cranial small intestine) and phosphorus (caudal small intestine). The endocrine function of the pancreas is performed by small groups of cells, the islets of Langerhans (Fig. 12-15), which are completely surrounded by acinar or exocrine cells that produce digestive enzymes. During embryonic development of the pancreas, a close relationship exists between the endocrine and exocrine portions. Evidence suggests that islet, acinar, and ductal cells arise from a common multipotential precursor cell. In early embryonic development, the endocrine cells are integrated within the exocrine matrix of the pancreatic bud. They subsequently accumulate in nonvascularized clusters and later become separated from the exocrine tissue and then independently vascularized. Fig. 12-15 Pancreatic islet, normal dog. The pancreatic islets of normal animals contain multiple types of cells. The predominant secretory cells are the β cells, which function in the biosynthesis of insulin but co-secrete islet amyloid polypeptide. The glucagon-secreting α cells are less numerous than β cells. δ Cells and F, or PP, cells in the islets secrete somatostatin and pancreatic polypeptide, respectively. The different cell types of the endocrine pancreatic cells can be differentiated by cytochemical and immunohistochemical techniques, and by electron microscopy (Web Fig. 12-4). The α, β, and δ cells have well-developed rough endoplasmic reticulum and Golgi complexes that participate in the biosynthesis of polypeptide hormones, as well as numerous secretory granules in the cytoplasm. Each type of endocrine cell in the pancreatic islets has secretory granules with distinct ultrastructural characteristics, which can be used to identify the cell types; however, immunohistochemical identification of the specific islet hormone is a more accurate method of identifying different cell types in the pancreatic islets. Web Fig. 12-4 Pancreatic islet, normal dog. Pathogenic Mechanisms of Endocrine Diseases Although injury of cells in endocrine glands is often attributable to processes, such as necrosis, inflammation, and autoimmunity, discussed in Chapters 1, 3, and 5, respectively, many diseases of endocrine glands are characterized by dramatic functional disturbances and characteristic clinicopathologic alterations affecting one or more body systems. The affected animal can have changes primarily involving the skin (alopecia caused by hypothyroidism), nervous system (seizures caused by hyperinsulinism), urinary system (polyuria caused by diabetes mellitus, diabetes insipidus, and hyperadrenocorticism), or skeletal system (fractures induced by hyperparathyroidism). There are several mechanisms that can disrupt normal endocrine function, with the majority of processes resulting in insufficient or excessive hormone production. Failure of development also results in primary hypofunction of an endocrine gland. The classic example of this mechanism in animals is the failure of oropharyngeal ectoderm to differentiate completely into trophic hormone–secreting cells of the adenohypophysis in dogs, resulting in pituitary dwarfism (see the section on Disorders of Dogs). In secondary hypofunction of an endocrine gland, a destructive lesion in one organ, such as the pituitary gland, interferes with the secretion of a trophic hormone. This results in hypofunction of the target endocrine gland. Large, endocrinologically inactive pituitary neoplasms in adult dogs, cats, and other animals can interfere with the secretion of multiple pituitary trophic hormones and result in clinically detectable hypofunction of the adrenal cortex (Fig. 12-16), follicular cells of the thyroid gland, and gonads. Fig. 12-16 Secondary hypofunction of adrenal glands, brain, pituitary gland and left (longitudinal section) and right (cross section) adrenal glands, dog. Primary hyperfunction of an endocrine gland is one of the most important pathologic mechanisms of endocrine disease in animals. The cells of a lesion, often a neoplasm derived from endocrine cells, autonomously synthesize and secrete a hormone at a rate in excess of the body’s ability to use and degrade it, thereby resulting in a syndrome caused by hormone excess. Examples are summarized in Table 12-1. TABLE 12-1 Primary Hyperfunction of an Endocrine Gland In this mechanism of endocrine disease, a lesion in one organ (e.g., adenohypophysis) secretes an excess of a trophic hormone that leads to long-term stimulation and hypersecretion of a hormone by a target organ. The classic example in animals is the ACTH-secreting neoplasm derived from pituitary corticotrophs in dogs and cats (Fig. 12-17). The functional disturbances and lesions are caused by increased blood cortisol concentrations resulting from the ACTH-stimulated hypertrophy and hyperplasia of the cells of the zonae fasciculata and reticularis of the adrenal cortex. In some aging dogs with notable adrenal cortical enlargement and functional disturbances of cortisol excess, no gross or histopathologic evidence of a neoplasm is present in the pituitary gland. These animals can have a change in negative feedback control as the result of an age-related increase in monoamine oxidase-β in the hypothalamus and increased metabolism of dopamine. The outcome is reduced inhibition of ACTH production by the pars intermedia of the pituitary gland leading to severe corticotroph hyperplasia, increased ACTH concentration in the blood, and long-term stimulation of the adrenal cortex, resulting in the syndrome of cortisol excess. Fig. 12-17 Secondary hyperfunction of adrenal glands, brain, pituitary gland and left and right adrenal glands, dog. Certain neoplasms of nonendocrine tissue in animals secrete either new humoral substances or hormones that share chemical and/or biologic characteristics with the “native” hormones secreted by an endocrine gland. Most of the recently discovered humoral substances secreted by nonendocrine neoplasms are peptides rather than steroids, iodothyronines, or catecholamines. The nonpeptide hormones require more complex biosynthetic pathways and are infrequently produced by cancer cells. Pseudohyperparathyroidism or humoral hypercalcemia of malignancy is a clinical syndrome primarily produced by the autonomous hypersecretion of PTH–related peptide (PTHrP) by cancer cells. PTHrP interacts with the parathyroid hormone receptor in target cells (e.g., bone and kidneys) and results in persistent, often life-threatening hypercalcemia. A well-characterized example of this disease mechanism in animals is the adenocarcinoma of the apocrine glands of the anal sac in dogs (see the section on Disorders of Dogs). These neoplasms produce PTHrP, which mimics the action of PTH and results in an accelerated mobilization of calcium from bone by osteoclasts leading to the development of persistent hypercalcemia. Serum PTH concentrations are lower in dogs with apocrine carcinomas than in controls, and PTH concentrations are undetectable in neoplastic tissue. The administration of an exogenous hormone can influence the activity of target cells either directly or indirectly and result in important functional disturbances. It is well recognized that the chronic administration of potent preparations of adrenal cortical steroids at inappropriately large daily doses (for the symptomatic treatment of various diseases) can produce most of the functional disturbances that are secondary to an endogenous hypersecretion of cortisol. Increased concentrations of exogenous cortisol result in notable atrophy of the adrenal cortex, particularly the ACTH-dependent zonae fasciculata and reticularis (Fig. 12-18). Similarly the administration of excessively large doses of insulin can result in hypoglycemia, and an excess of T4 or T3 can result in hyperthyroidism, especially in certain species, such as cats, which have limited capacities to conjugate T4 with glucuronic acid and thus enhance its biliary excretion. Fig. 12-18 Iatrogenic hyperadrenocorticism, left and right adrenal glands, dog. The administration of progestogens to dogs indirectly results in a syndrome of growth hormone excess. For example, the injection of medroxyprogesterone acetate for the prevention of estrus in dogs stimulates the expression of the growth hormone gene in the mammary glands and results in elevated circulating growth hormone concentrations, producing many of the clinical manifestations of acromegaly. The excessive skinfolds (Fig. 12-19), expansion of interdigital spaces, and abdominal enlargement in dogs with iatrogenic acromegaly are related to the protein anabolic effects of growth hormone. Elongation of bones in response to exogenous progestogens requires functional growth plates and osteogenic surfaces. Fig. 12-19 Iatrogenic acromegaly, beagle (center) compared with unaffected littermates (left and right). The primary defense mechanism employed by the endocrine system is the hierarchy of hormonal regulation known as the hypothalamic-pituitary-target gland axis (see Fig. 12-3). The hypothalamus produces a number of releasing and inhibitory hormones in response to sensory input from the central nervous system (CNS). These releasing and inhibitory hormones act on anterior or posterior portions of the pituitary gland to stimulate or prevent the release of trophic hormones. Trophic hormones act on specific endocrine glands, stimulating them to produce hormones that exert ultimate actions on downstream tissues. Under normal circumstances, the action of a hormone is self-limiting because of the existence of negative feedback loops for each hormone series in which secretion of a particular hormone ultimately leads to inhibition of its subsequent secretion. Negative feedback from target endocrine glands can be directed at the hypothalamus, the pituitary gland, or both. Disorders known or thought to have a genetic basis and/or be inherited are listed in Web Table 12-1. WEB TABLE 12-1 Inherited Endocrine Diseases of Animals Hypopituitarism and Neoplasms of the Adenohypophysis Aplasia and Prolonged Gestation: See the section on Disorders of Ruminants for a discussion of aplasia and prolonged gestation. Pituitary Cysts and Pituitary Dwarfism: See the section on Disorders of Dogs for a discussion of pituitary cysts and pituitary dwarfism. Endocrinologically Inactive Chromophobe Adenomas: Nonfunctional pituitary neoplasms occur in dogs and cats but are uncommon in other species. Although chromophobe adenomas seem endocrinologically inactive, they can cause significant functional disturbances and clinical signs by compressing and causing atrophy of adjacent portions of the pituitary gland, as well as dorsal extension into the overlying brain (Fig. 12-20). The clinical disturbances result either from the lack of secreted pituitary trophic hormones and subsequent diminished target organ function (e.g., adrenal cortex; see Fig. 12-16) or from dysfunction of the CNS. Affected animals often have decreased spontaneous activity, incoordination, and disturbances of balance; are weak; and sometimes collapse after exercise. Chronically affected animals are blind and have dilated and fixed pupils because of compression and disruption of the optic nerves by dorsal extension of the pituitary neoplasms (see Fig. 12-20). Endocrinologically inactive pituitary adenomas often become large before they cause clinical signs or kill the animal (see Figs. 12-16 and 12-20). Fig. 12-20 Adenoma, pituitary gland, dog. Clinical signs reported with nonfunctional pituitary adenomas and hypopituitarism are not specific and could be confused with other disorders of the CNS, such as brain neoplasms and encephalitis, or with chronic renal disease. There is no effect on body stature secondary to compression of the pars distalis and interference with growth hormone secretion because these neoplasms usually arise in adult animals that have already completed their growth. However, atrophy of the skin and loss of muscle mass could be related in part to a lack of the protein anabolic effects of growth hormone. Impaired secretion of pituitary trophic hormones often leads to a reduced basal metabolic rate because of decreased TSH secretion and hypoglycemia secondary to trophic atrophy of the adrenal cortex (see Fig. 12-16). Pituitary Gland Carcinomas: Pituitary gland carcinomas are uncommon neoplasms compared with adenomas but have been seen in older dogs and cattle. They usually are endocrinologically inactive but can cause significant functional disturbances by destroying the pars distalis and neurohypophyseal system, leading to panhypopituitarism and diabetes insipidus. Carcinomas are large and invade extensively into the overlying brain, along the ventral aspect of the cranial cavity, and into the basisphenoid bone where they cause osteolysis. Metastases occur infrequently to cervical lymph nodes or distant sites such as the spleen or liver. Carcinomas are highly cellular and often have large areas of hemorrhage and necrosis. Giant cells, nuclear pleomorphism, and mitotic figures are encountered more frequently than in adenomas. Craniopharyngiomas (Intracranial Germ Cell Tumors): Craniopharyngiomas are benign neoplasms derived from epithelial remnants of the oropharyngeal ectoderm of the craniopharyngeal duct (Rathke’s pouch). They often occur in animals younger than those with other types of pituitary neoplasms and are present in either suprasellar or infrasellar locations. Craniopharyngioma is associated with dwarfism in young dogs because it causes subnormal secretion of somatotrophin and other trophic hormones at an early age, before closure of the growth plates. Craniopharyngiomas and suprasellar germ cell tumors often are large and grow along the ventral aspect of the brain, where they can surround several cranial nerves. In addition, they extend dorsally into the hypothalamus and thalamus (Fig. 12-21). The resulting clinical signs often occur because of a combination of the following: Fig. 12-21 Craniopharyngioma (C), pituitary area, left and right adrenal glands, left and right thyroid glands, dog. • A lack of secretion of pituitary trophic hormones resulting in trophic atrophy and subnormal function of the adrenal cortex and thyroid gland, atrophy of the gonads, and failure to attain somatic maturation because of a lack of secretion of growth hormone • Disturbances in water metabolism (polyuria, polydipsia, low urine specific gravity, and osmolality) resulting from an interference in the synthesis and release of ADH by the large neoplasm • Deficits in cranial nerve function • CNS dysfunction caused by extension into the overlying brain Pars Intermedia Adenomas: Adenomas derived from cells of the pars intermedia are the most common type of pituitary gland neoplasm in horses and the second most common type in dogs, but they are rare in other species. Adenomas develop in older horses, more frequently in females. Nonbrachycephalic breeds of dogs have adenomas of the pars intermedia more often than brachycephalic breeds. The clinical syndrome reported with neoplasms of the pars intermedia in horses is characterized by polyuria, polydipsia, polyphagia, muscle weakness, somnolence, intermittent hyperpyrexia, and generalized hyperhidrosis. The affected horses often develop hirsutism because of a failure to seasonally shed hair. The hair over most of the trunk and extremities is long (up to 10 to 12 cm), abnormally thick and wavy, and often matted (Fig. 12-22). Horses with larger neoplasms sometimes have insulin-resistant hyperglycemia and glycosuria, probably resulting from the downregulation of insulin receptors on target cells induced by chronic excessive intake of feed and hyperinsulinemia. The disturbances in carbohydrate metabolism, ravenous appetite, hypertrichosis, and hyperhidrosis reflect deranged hypothalamic function caused by compression of the overlying hypothalamus by the large pituitary neoplasms. The hypothalamus is the primary center for homeostatic regulation of body temperature, appetite, and cyclic shedding of hair. Fig. 12-22 Hirsutism, skin, horse. In horses, adenomas of the pars intermedia often are large neoplasms that extend out of the fossa hypophysialis and severely compress the overlying hypothalamus (Fig. 12-23). The adenomas are yellow to white and multinodular and enclose the pars nervosa. When the neoplasm is incised, the pars distalis usually can be identified as a compressed subcapsular rim of tissue on the anterior margin. A sharp line of demarcation remains between the neoplasm and the compressed pars distalis. The neoplastic cells are arranged in cords and nests along the capillaries and connective tissue septae and are large, cylindrical, spindle-shaped, or polyhedral with oval hyperchromatic nuclei (Fig. 12-24). The pattern is often reminiscent of that of the prominent pars intermedia of normal horses. Ribbons of more cuboidal to columnar neoplastic cells occasionally form follicular structures that have dense eosinophilic colloid.
Endocrine System*
Structure and Function
Pituitary Gland (Hypophysis)
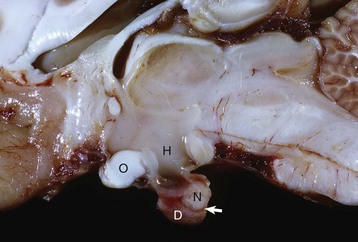
Longitudinal section of the pituitary region illustrating the close relationship to the optic chiasm (O), hypothalamus (H), and overlying brain. The pars distalis (D) forms a major part of the adenohypophysis and completely surrounds the pars nervosa (N). The residual lumen of Rathke’s pouch (arrow) separates the pars distalis and pars nervosa and is lined by the pars intermedia. (Courtesy Dr. C. Capen, College of Veterinary Medicine, The Ohio State University.)
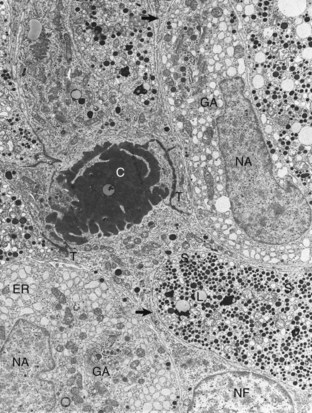
Follicular cells (NF) in the pars distalis form a framework and extend cytoplasmic processes (arrows) around extracellular accumulations of colloid (C). Adjacent follicular cells are joined by prominent terminal bars (T). Acidophils in the storage phase of the secretory cycle contain numerous large, uniformly electron-dense secretory granules (S), scattered lipofuscin (L) bodies, a small amount of endoplasmic reticulum (ER), and a small Golgi apparatus. Hypertrophied acidophils (NA) have few mature secretory granules but many distended profiles of endoplasmic reticulum and large Golgi apparatuses (GA) associated with prosecretory granules in the process of formation. TEM. Uranyl acetate and lead citrate stain. (Courtesy Dr. C. Capen, College of Veterinary Medicine, The Ohio State University.)
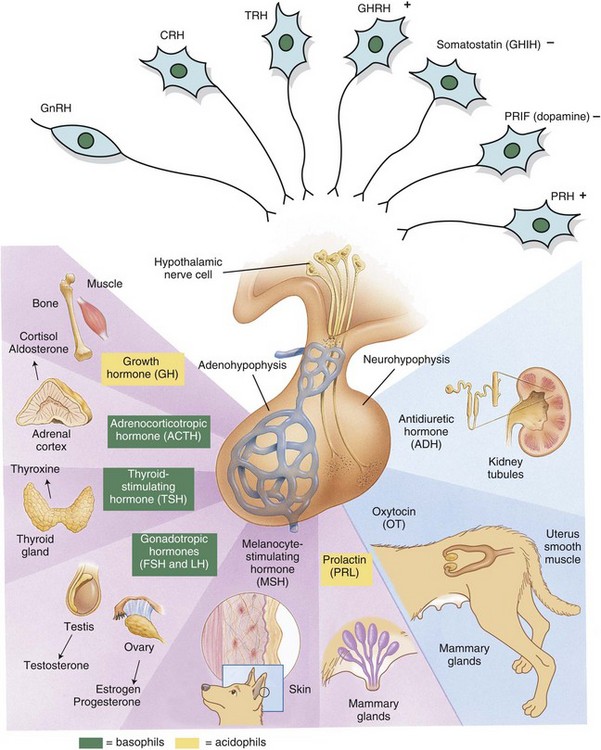
Releasing hormones produced by the hypothalamus act on anterior or posterior portions of the pituitary gland to release trophic hormones. Trophic hormones act on specific endocrine glands, stimulating them to produce hormones that exert ultimate actions on downstream tissues. CRH, Corticotropin-releasing hormone; FSH, follicle-stimulating hormone; GHIH, growth hormone–inhibiting hormone; GHRH, growth hormone–releasing hormone; GnRH, gonadotropin-releasing hormone; LH, luteinizing hormone; PRH, prolactin-releasing hormone; PRIF, prolactin release–inhibiting factor; TRH, thyrotropin-releasing hormone. (Modified from Huether SE, McCance KL: Understanding pathophysiology, ed 2, St Louis, 2000, Mosby; and Squire L, Bloom F, McConnell S: Fundamental neuroscience, ed 2, San Diego, 2003, Academic Press.)
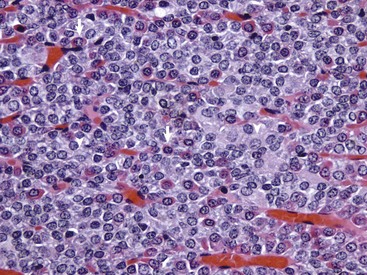
The pars distalis is composed of acidophils (arrows), basophils (none shown here) and chromophobes (arrowheads). H&E stain. (Courtesy Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois.)
Adrenal Gland
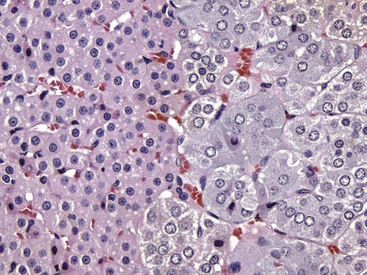
Interface between the finely vacuolated (lipid droplets) cells of the adrenocortical zona reticularis (left) and chromaffin cells of the adrenal medulla (right). H&E stain. (Courtesy Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois.)
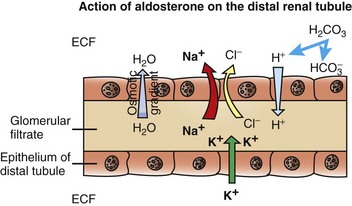
The resulting osmotic gradient facilitates movement of water from the glomerular filtrate into the extracellular fluid (ECF). (Redrawn with permission from Dr. C. Capen, College of Veterinary Medicine, The Ohio State University.)
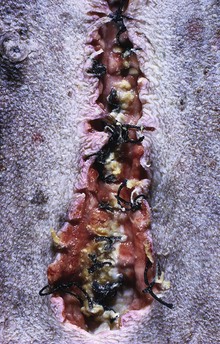
Wounds heal slowly in dogs with cortisol excess because of an inhibition of fibroblastic proliferation. (Courtesy Dr. C. Capen, College of Veterinary Medicine, The Ohio State University.)
Thyroid Gland
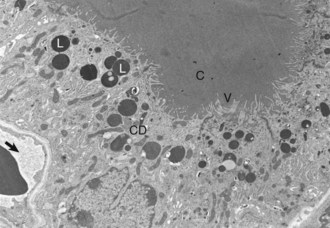
Thyroid follicular cells with long microvilli (V) that extend into the colloid (C) within the follicular lumen. Numerous lysosomes (L) and colloid droplets (CD) are present in the apical portion of the follicular cells. An interfollicular capillary (arrow) is present at the base of the follicle. TEM. Uranyl acetate and lead citrate stain. (Courtesy Dr. C. Capen, College of Veterinary Medicine, The Ohio State University.)
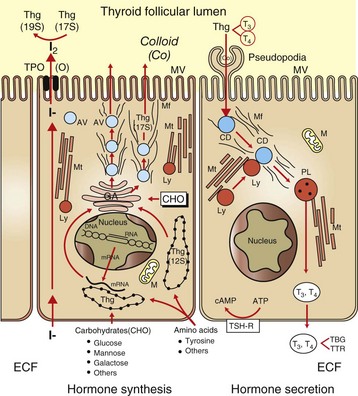
Raw materials, such as iodide, are concentrated by follicular cells and rapidly transported into the lumen (left side of drawing). Amino acids (tyrosine and others) and sugars are assembled by follicular cells into thyroglobulin (Thg), packaged into apical vesicles (AV) and released into the lumen. The iodination of tyrosyl residues with the thyroglobulin molecule to form thyroid hormones occurs within the follicular lumen. Elongation of microvilli (MV) and endocytosis of colloid (Co) by follicular cells occur in response to thyroid-stimulating hormone (TSH) stimulation (right side of drawing). The intracellular colloid droplets (CD) fuse with lysosomal bodies (Ly), active thyroid hormone is enzymatically cleaved from thyroglobulin, and free T4 and T3 are released into the circulation. ATP, Adenosine triphosphate; cAMP, cyclic adenosine monophosphate; CHO, carbohydrates; ECF, extracellular fluid; GA, Golgi apparatus; M, mitochondrion; Mf, microfilaments; Mt, microtubules; PL, phagolysosome; TBG, thyroid-binding globulin; TPO, thyroid peroxidase; TSH-R, thyroid-stimulating hormone receptor; TTR, transthyretin. (From Capen CC: Pathophysiology of the thyroid gland. In Dunlop RH, Malbert C-H, editors: Veterinary pathophysiology, Ames, IA, 2004, Blackwell Publishing.)
Follicular epithelial cells following prolonged thyroid-stimulating hormone stimulation are columnar. Note the many collapsed follicles. The lumens of remaining follicles contain pale pink colloid and have numerous endocytic vacuoles at the epithelial cell-follicular lumen interface. H&E stain. (Courtesy Dr. B. Harmon, College of Veterinary Medicine, The University of Georgia; and Noah’s Arkive, College of Veterinary Medicine, The University of Georgia.)
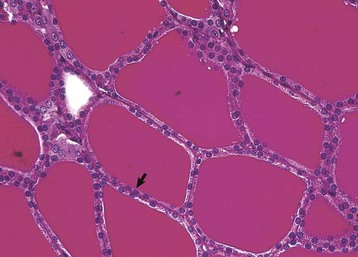
Thyroid follicular epithelial cells (arrow) after long-term administration of exogenous thyroxine are cuboidal and follicular lumens are distended with dense colloid. Periodic acid–Schiff reaction. (Courtesy Dr. C. Capen, College of Veterinary Medicine, The Ohio State University.)
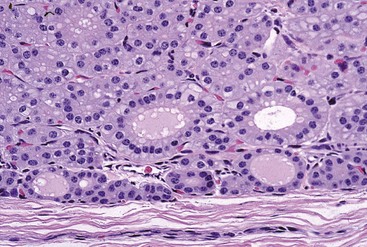
Thyroid C (Parafollicular) Cells
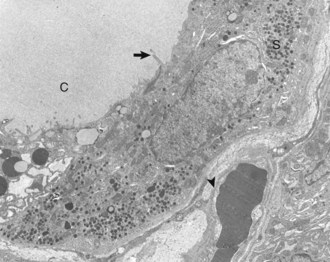
Thyroid C (parafollicular) cell with numerous secretory granules (S) and moderate development of Golgi apparatus and rough endoplasmic reticulum. Microvilli from follicular cells (arrow) extend into the colloid of the follicular lumen (C). The secretory polarity of the C cell is directed toward an interfollicular capillary (arrowhead) with fenestrae. TEM. Uranyl acetate and lead citrate stain. (Courtesy Dr. C. Capen, College of Veterinary Medicine, The Ohio State University.)
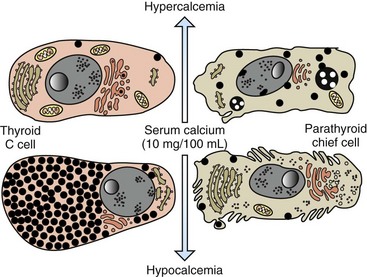
C cells accumulate secretory granules in response to hypocalcemia, whereas chief cells are nearly degranulated but have an increased development of synthetic and secretory organelles. In response to hypercalcemia, C cells are degranulated and parathyroid chief cells are predominantly in the inactive stage of the secretory cycle. (Redrawn with permission from Dr. C. Capen, College of Veterinary Medicine, The Ohio State University.)
Parathyroid Glands
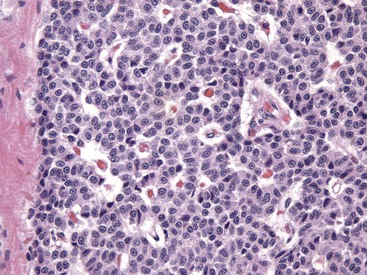
Numerous chief cells are separated and supported by a fine fibrovascular stroma. H&E stain. (Courtesy Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois.)
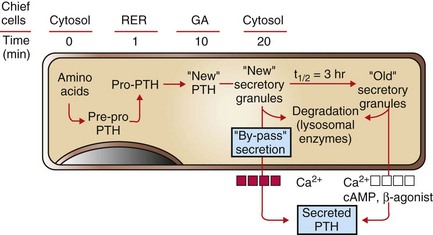
Recently synthesized and processed active PTH can be released directly without entering the storage pool of mature (“old”) secretory granules in the cytoplasm of chief cells. PTH from the storage pool can be mobilized by cyclic adenosine monophosphate (cAMP) and β-agonists, such as epinephrine, norepinephrine, and isoproterenol, and by lowered blood calcium ion, whereas secretion from the pool of recently synthesized PTH can be stimulated only by a decreased calcium ion concentration. RER, Rough endoplasmic reticulum; GA, Golgi apparatus. (Redrawn with permission from Dr. C. Capen, College of Veterinary Medicine, The Ohio State University.)
Pancreatic Islets
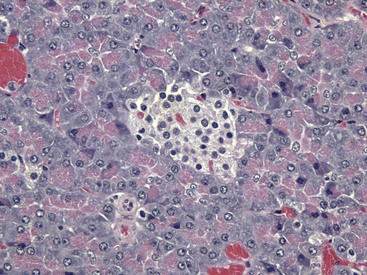
The islet is surrounded by the exocrine pancreas. H&E stain. (Courtesy Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois.)
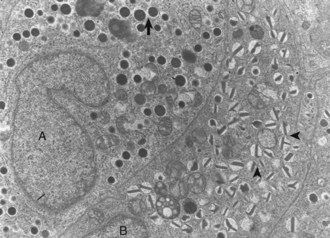
Differences in secretion granules between β cells (B) and α cells (A); the internal cores of secretion granules in insulin-secreting β cells (arrowheads) are bar- or Y-shaped, with a prominent space between the limiting membrane and internal core. Secretion granules of the glucagon-secreting α cells have an electron-dense, circular, internal core with a narrow submembranous space (arrow). TEM. Uranyl acetate and lead citrate stain. (Courtesy Dr. C. Capen, College of Veterinary Medicine, The Ohio State University.)
Responses to Injury
Primary Hypofunction of an Endocrine Gland
Secondary Hypofunction of an Endocrine Gland
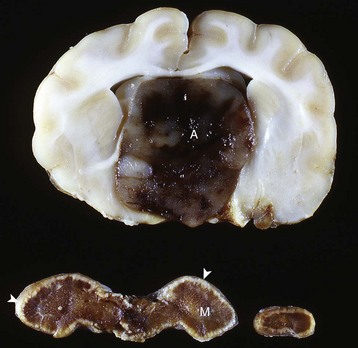
A large nonfunctional chromophobe adenoma (A) has invaded and completely destroyed the adenohypophysis and hypothalamus, and infiltrated into the thalamus. Destruction of the adenohypophysis has resulted in a lack of secretion of thyrotropin, adrenocorticotropin, and other pituitary trophic hormones, resulting in severe bilateral (symmetrical) trophic atrophy of the adrenal cortex (arrowheads), especially the adrenocorticotropic hormone–dependent zona fasciculata and zona reticularis, and consequently, in a relatively more prominent medulla (M). (Courtesy Dr. C. Capen, College of Veterinary Medicine, The Ohio State University.)
Primary Hyperfunction of an Endocrine Gland
Neoplasia
Hormone
Lesion/Sign
Acidophil adenoma (pituitary gland)
Growth hormone
Acromegaly
Adrenal cortical adenoma/carcinoma
Estrogen
Feminization
Pheochromocytoma (adrenal medulla)
Norepinephrine
Hypertension
Thyroid follicular cell adenoma
T4, T3
↑Basal metabolic rate
C-cell adenoma/carcinoma (thyroid gland)
Calcitonin
Osteosclerosis
Parathyroid gland chief cell adenoma
Parathyroid hormone
Fibrous osteodystrophy
Pancreatic β-cell adenoma/carcinoma
Insulin
Hypoglycemia
Secondary Hyperfunction of an Endocrine Gland
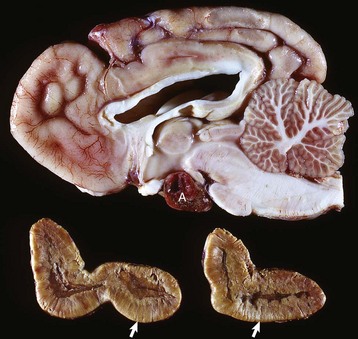
Corticotroph (adrenocorticotropic hormone [ACTH]-secreting) chromophobe adenoma (A) in the pituitary gland and bilateral (symmetrical) enlargement of the adrenal glands. The chronic secretion of ACTH has resulted in bilateral (symmetrical) hypertrophy and hyperplasia of secretory cells of the zona fasciculata and zona reticularis in the adrenal cortex (arrows) and excessive secretion of cortisol. (Courtesy Dr. C. Capen, College of Veterinary Medicine, The Ohio State University.)
Hypersecretion of Hormones or Hormonelike Factors by Nonendocrine Neoplasms
Iatrogenic Syndromes of Hormone Excess
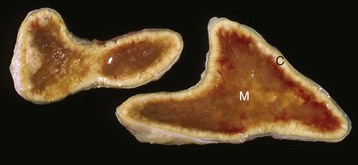
Hyperadrenocorticism, caused by long-term administration of exogenous corticosteroids, has resulted in notable trophic atrophy of the adrenocorticotropic hormone–dependent zona fasciculata and zona reticularis of the adrenal cortex (C). The adrenal medulla (M) comprises a relatively greater percentage of the atrophic adrenal gland than of a normal adrenal gland. (Courtesy Dr. C. Capen, College of Veterinary Medicine, The Ohio State University.)

Note the coarseness of the facial features and the markedly thick folds of the skin of the face. These characteristic changes are the result of the protein anabolic effects of somatotropin (produced by hyperplastic mammary ductular epithelial cells), which have been stimulated by the administration of exogenous medroxyprogesterone acetate. (Courtesy Dr. P. Concannon, College of Veterinary Medicine, Cornell University.)
Defense Mechanisms
Disorders of Domestic Animals
Condition
Species/Breed
Pattern of Inheritance
Adenohypophyseal aplasia
Jersey and Guernsey cattle
Unknown
Pituitary dwarfism
German shepherds
Autosomal recessive
Hypoadrenocorticism
Bearded collies, Nova Scotia duck tolling retriever, Portuguese water dogs, standard poodles
Unknown or
autosomal recessive
Adrenal hyperplasia-like syndrome
Chow Chows, Pomeranians, poodles, Samoyeds
Unknown
Dyshormonogenetic goiter
Abyssinian cats, Afrikaner cattle, rat and toy fox terriers, Saanen dwarf goats, sheep
Autosomal recessive
Disorders of the Adenohypophysis
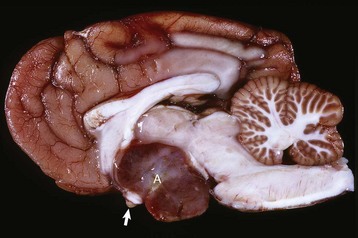
A large pituitary adenoma (A) has extended dorsally and compresses the overlying brain. The optic chiasm (arrow) is also severely compressed. The adenohypophysis, neurohypophysis, and hypothalamus have been destroyed by the neoplasm. (Courtesy Dr. C. Capen, College of Veterinary Medicine, The Ohio State University.)
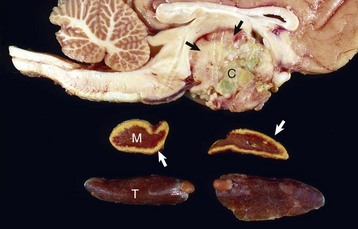
The neoplasm has extended dorsally through the hypothalamus and compressed the thalamus (black arrows). The neoplasm has also destroyed the adenohypophysis and neurohypophysis, resulting in severe bilateral (symmetrical) trophic atrophy of the adrenal cortices (white arrows). The adrenal glands consist predominantly of medulla (M) surrounded by a thin rim of cortex (capsule plus zona glomerulosa). Although the thyroid follicular cells are atrophic, the overall gland (T) size is within normal limits because of colloid involution of the follicles. (Courtesy Dr. C. Capen, College of Veterinary Medicine, The Ohio State University.)
Hyperpituitarism and Neoplasms of the Adenohypophysis

The hirsutism is the result of a failure to shed hair because of hypothalamic compression by an adenoma of the pars intermedia. (Courtesy Dr. C. Capen, College of Veterinary Medicine, The Ohio State University.)
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree