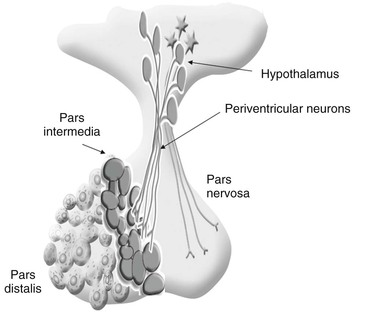
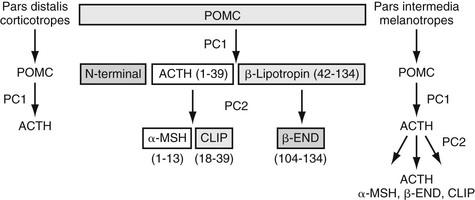
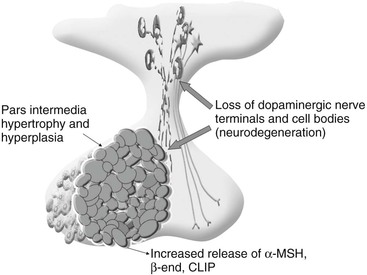
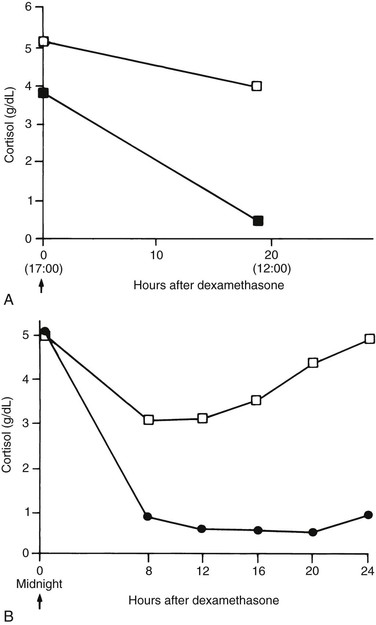
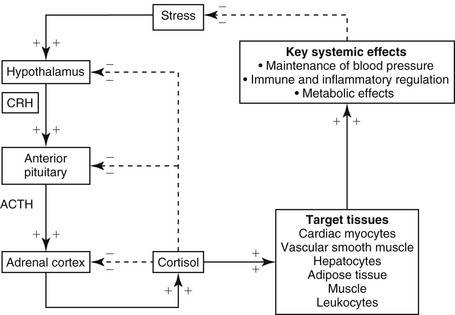
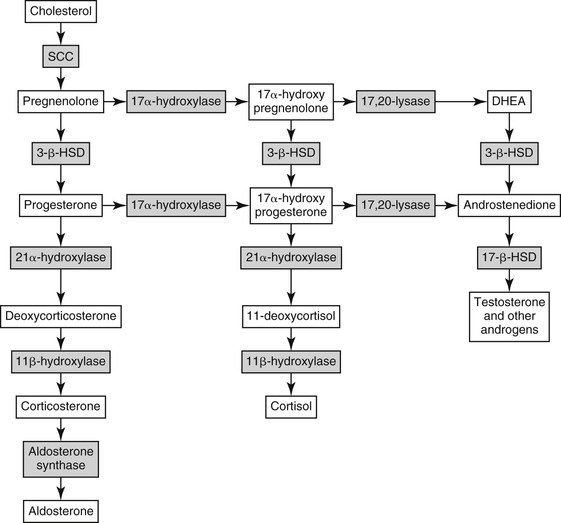
dianne mcfarlane, sherrill a. fleming, Consulting Editors ▪ Equine Endocrine and Metabolic Disorders Dianne McFarlane, Consulting Editor Dianne McFarlane The pituitary gland is composed of two embryologically distinct portions: an adenohypophysis, derived from invagination of the pharyngeal epithelium known as Rathke’s pouch, and the neurohypophysis, derived from neural tissue of the hypothalamus. The adenohypophysis can be further divided into the pars distalis, pars tuberalis, and pars intermedia. The pars distalis contains five different endocrine cell types, each of which is responsible for the release of a unique hormone or set of hormones in response to hypothalamic releasing factors delivered from the median eminence via the hypothalamic-hypophyseal portal system. The pars tuberalis is a highly vascular band of cells surrounding the pituitary stalk. The pars tuberalis is rich in melatonin receptors and is believed to be involved in regulating the seasonal rhythm of reproductive hormone production.1 The neurohypophysis or pars nervosa is a collection of nerve axons and terminals that originate in the paraventricular and supraoptic nuclei of the hypothalamus. Oxytocin and arginine vasopression (antidiuretic hormone) produced in the cell bodies of these nuclei are transported into the pars nervosa for storage and eventual release into the systemic circulation. The pars intermedia of the horse incorporates tissue derived from both the adeno- and neurohypophysis.2 It is composed of a single endocrine cell type, the melanotrope. Melanotropes are directly innervated by nerve terminals of the hypothalamic periventricular dopaminergic neurons. These originate in the periventricular nucleus of the hypothalamus adjacent to the third ventricle, project through the infundibulum, and terminate in the pars intermedia (Fig. 41-1).3 These neurons release the neurotransmitter dopamine, which acts to tonically inhibit the release of hormones from adjacent melanotropes. Dopamine released from the nerve terminals interacts at dopamine (D2) receptors on the melanotropes to inhibit cell proliferation, transcription of proopiomelanocortin (POMC), and release of POMC-derived peptides.4 Additional regulatory signals to the pars intermedia may be delivered by direct systemic arterial supply and from the hypothalamic-hypophyseal portal veins.5 In addition to being under tonic inhibition, melanotropes are also positively regulated by thyrotropin-releasing hormone (TRH), which stimulates hormone release from melanotropes.6 Additional regulatory molecules likely exist but at this time are poorly defined in the horse. The primary product of the melanotrope is the hormone precursor protein POMC, which is also expressed by the corticotropes of the pars distalis. However, due to differential post-translational processing by proteases called prohormone convertases, each cell type secretes a different complement of POMC-derived peptides (Fig. 41-2). Due to the action of prohormone convertase I, POMC in corticotropes is primarily processed into adrenocorticotropic hormone (ACTH). ACTH circulates to the adrenal cortex, where it stimulates secretion of cortisol. Melanotropes contain active prohormone convertase I and II, and therefore POMC in the pars intermedia (PI) is cleaved into the secretory peptides, α-melanocyte stimulating hormone (α-MSH), β-endorphin (β-end), and corticotropin-like intermediate lobe peptide (CLIP). A small amount of ACTH may also be produced. Further processing of the peptides, including cleavage of C-terminal amino acids and N-acetylation, serves to control the activity of the final peptide product. For example, the most abundant form of β-end produced in the normal horse’s pars intermedia is Ac-β-end (1-27), which lacks opioid activity. The most abundant β-end in horses with pituitary pars intermedia dysfunction (PPID) is β-end (1-31), which is an opioid agonist.7 The physiologic role of the pars intermedia POMC-derived peptides, α-MSH, β-end, and CLIP, has not been extensively studied in the horse. In other species, α-MSH has several diverse actions, which are mediated through interaction with one of five distinct G-protein–coupled melanocortin receptors. α-Melanocyte stimulating hormone is named such due to its ability to induce skin pigmentation in amphibians. Its role in pigmentation is through interaction with melanocortin receptor 1 (MC1R), which is predominantly expressed in skin. In horses, mutation of the MC1R gene is associated with the chestnut coat color.8 In white Camarque horses the degree of coat pigmentation is directly correlated to the plasma concentration of α-MSH.9 α-MSH is also an integral mediator in control of energy homeostasis. MC3R and MC4R are both expressed in the central nervous system (CNS), particularly in the hypothalamus, where they function in the leptin-melanocortin pathway, regulating appetite-satiety balance and fat metabolism.10 Animals and humans lacking functional MC3R or MC4R are obese, and melanocortin receptor defects are a common monogenetic cause of obesity in humans.11 Plasma α-MSH concentration in obese men has been reported to be higher than in lean men.12 It has been suggested high plasma concentration in obese individuals may be an attempt to maintain homeostasis in individuals with a defect in MC4R. Plasma α-MSH concentration was also found to be positively correlated to obesity in horses.13Another function of α-MSH is as a potent antiinflammatory agent.14 α-MSH has been demonstrated to have multiple immune-modulating effects. Its most profound effect is in regulation of cytokine response. α-MSH inhibits activation of NF-κB by lipopolysaccharide (LPS) and interferon-γ (IFN-γ). Recent data in mice have suggested that α-MSH may suppress LPS-mediated inflammation by facilitating the interaction of IRAK and IRAK-M.15 IRAK is a kinase that functions in activation of NF-κB following TLR-4 (the receptor for bacterial LPS) stimulation.15 IRAK-M is a negative regulator of IRAK. As a result, NF-κB activation and proinflammatory cytokine release of tumor necrosis factor (TNF)-α, interleukin (IL)-1β, and IL-6 are all decreased following α-MSH administration. Fever and other clinical evidence of inflammation are also reduced. In addition, α-MSH also inhibits neutrophil function, including adhesion, chemotaxis, and oxidative burst.16–17 β-End is a known endogenous opioid. Secretion of β-end provides analgesia and behavioral modification. It also suppresses immune responsiveness and has effects on vascular tone.18–19 Corticotropin-like intermediate lobe peptide (CLIP, ACTH 18-34) has not been extensively studied in any species. In pancreatic islet cells in culture CLIP was shown to be a pancreatic β-cell secretagogue, stimulating the release of insulin.20 However, when administered to rats by either intraperitoneal or intraventricular injection, ACTH but not CLIP resulted in release of insulin and decrease in blood glucose.21 A distinct seasonal effect on the activity of the pars intermedia of horses and ponies has been demonstrated in horses residing in tropical and temperate environments.22–28 Plasma α-MSH concentration is considerably higher in horses and ponies in August through October, compared with samples collected in the winter, spring, and early summer. An effect of season on α-MSH concentration has been described for humans, hamsters, and sheep.29–31 The functional importance of the seasonal cycle is unknown, but several physiologic events occur in parallel with the α-MSH cycle. In sheep, body weight, voluntary food intake, and condition all peak simultaneously with α-MSH, with seasonal maximums occurring in September. Soay sheep with surgically created hypothalamic-pituitary disconnection have an increase in circulating concentration of α-MSH and chronic increase in body weight.30 These findings suggest that α-MSH or other POMC-derived peptides may play a role in metabolic preparation for winter in sheep. It is possible that horses and ponies have a seasonal increase in POMC-derived peptides to metabolically prepare them for a decrease in accessible food observed in the wild in winter. If so, dysregulation of this pathway might be associated with abnormalities in body weight and fat storage. Weight loss and abnormal fat distribution are two clinical signs associated with equine PPID. Development of a winter coat also begins as length of day decreases in the fall. The development of hypertrichosis in horses with PPID leads one to speculate that the naturally occurring seasonal increase in POMC-derived peptides contributes to development of winter coat growth. This has not been critically assessed in equids. Equine PPID is one of the most common diseases of horses and ponies 15 years and older.32–34 PPID was originally termed equine Cushing disease because of features similar to human Cushing disease. However, in contrast to human Cushing disease, PPID affects the pituitary pars intermedia rather than the pars distalis, is typically not a neoplastic condition, and the adrenocortical contribution to the clinical syndrome is of much less importance. To avoid confusion, equine Cushing disease is now more correctly referred to as PPID. In the past 2 decades the population of aged horses has increased dramatically. This, in conjunction with the vast amount of information available to the horse-owning public, has led to a heightened client awareness of age-associated equine health issues and a desire to promote healthy aging in their horses. As a result, diagnostic testing and treatment of horses for PPID has increased. Yet despite increased clinical recognition of this disease, much about PPID remains poorly understood. The pathologic hallmark of PPID is hypertrophy, hyperplasia, and microadenoma or macroadenoma formation in the pituitary pars intermedia with increased secretion of POMC peptides. Horses with PPID develop enlarged pituitaries that may reach five times normal weight. As the pars intermedia expands, it compresses the adjacent pituitary lobes and hypothalamus, often resulting in a loss of function of these tissues. In contrast, the pars intermedia remains active in horses with PPID, secreting relatively large quantities of POMC-derived peptides into the peripheral circulation. Horses with disease may have as much as a 40-fold increase in plasma concentration of pars intermedia POMC-derived peptides.35 Clinical signs of disease likely result from a combination of increased circulating POMC peptides and loss of neuroendocrine function of adjacent tissues. Evidence indicates loss of dopamine inhibition is critical in the pathology of PPID. Dopamine and dopamine metabolite concentrations in the pars intermedia of PPID horses are decreased eightfold compared with age-matched controls.7 Systemic supplementation of dopamine or a dopamine agonist to horses with PPID results in a decrease in plasma concentration of POMC peptides.35 Horses treated with the dopamine agonist pergolide show improvement in both clinical signs and biochemical abnormalities associated with disease.36–39 Immunohistochemistry of formalin-fixed tissue showed a fivefold decrease in pituitary dopaminergic nerve terminals (P < 0.001) and a 50% reduction in the number of dopaminergic periventricular cell bodies (P < 0.01) in the hypothalamus of PPID animals.40 Considered together, this evidence suggests a loss of functional periventricular dopaminergic neurons or “dopaminergic neurodegeneration” occurs in horses with PPID (Fig. 41-3). Loss of periventricular dopaminergic inhibition of the pars intermedia in other species results in pathologic changes similar to those of PPID. Surgical disruption of the periventricular hypophyseal dopaminergic tracts in rats results in increased expression of pars intermedia melanotropes.4 In addition, D2 dopamine receptor knockout mice develop pars intermedia lesions similar to PPID.41 These data suggest PPID is primarily a disease of hypothalamic origin, rather than the consequence of a spontaneously forming pituitary adenoma. One potential cause for dopaminergic neurodegeneration is oxidative stress. Oxidative stress causes modification of cellular components including proteins, DNA, and cell membrane lipids due to excessive exposure to reactive oxygen species leading ultimately to cell death, or in the case of neurons, neurodegeneration. Dopaminergic neurons are particularly vulnerable to oxidative damage because dopamine metabolism itself produces free radicals. Horses with PPID have evidence of oxidative damage, including accumulation of pars intermedia 3-nitrotyrosine40 and decreased plasma thiol.42 The oxidative damage in PPID does not appear to be the result of impaired antioxidant capacity because systemic and pituitary antioxidant capacity of affected horses appears unchanged.43 Equine PPID affects many aged equids, resulting in a variety of clinical signs including hypertrichosis (hirsutism), laminitis, muscle atrophy, fat accumulation, polydipsia, polyuria, secondary infections, lethargy, infertility, persistent lactation, sweating dysregulation, and metabolic abnormalities including hyperglycemia and hyperinsulinemia (Fig. 41-4).44–45 The most unique clinical manifestation of PPID is an abnormally long, curly haircoat that fails to shed (previously referred to as equine hirsutism, although more correctly called hypertrichosis). Often, horse owners may report the horse sheds its winter coat slowly or incompletely in the year(s) before development of a full failure to shed. Hair may be initially retained along the legs or under the mandible (Fig. 41-5). The mechanism responsible for development of hypertrichosis in horses with PPID has not been reported. The onset of hypertrichosis in an aged horse or pony is considered essentially pathognomonic for PPID, although the author has observed similar haircoats in severely debilitated aged animals with confirmed normal pituitary pars intermedia function. Laminitis secondary to PPID is reported to occur in approximately 30% to 40% of diagnosed cases and can necessitate euthanasia in affected animals.39,46–48 When adult horses with laminitis of unknown origin were examined, 30% to 70% had evidence of PPID,49–50 suggesting PPID may be underdiagnosed, especially when laminitis is the presenting complaint and hirsutism is absent. The pathogenesis of laminitis in PPID is not currently well understood but is the subject of ongoing investigation. Historically, it has been suggested that laminitis secondary to PPID is the consequence of high circulating cortisol concentration. However, the inability to induce experimental laminitis in normal horses using corticosteroids and the absence of hypercortisolemia or adrenal hyperplasia in most horses with PPID indicates this is unlikely to be the pathogenic mechanism. More recently, a primary role for hyperinsulinemia in induction of endocrinopathic laminitis has been proposed. Several lines of evidence support this hypothesis. First, sustained administration of a high concentration of insulin or dextrose to normal horses and ponies has been shown to induce laminitis that is similar both clinically and histologically to naturally occurring endocrinopathic laminitis.51–54 Additionally, several investigators have documented a strong association between serum insulin concentration and naturally occurring laminitis.50,55–56 In the author’s experience weight loss due to muscle mass atrophy is the most common and possibly earliest clinical sign of PPID (Fig. 41-6). Despite weight loss, PPID horses often have a potbellied appearance and, as a consequence, owners may fail to notice the lost weight. Weight loss and muscle atrophy may result from several factors, including poor dentition, poor nutrition, heavy parasite burden, minimal exercise, and protein catabolism induced by increased cortisol activity. Histologic evidence of type 2 myofiber atrophy has been documented in muscle biopsies from horses with PPID, consistent with corticosteroid-associated muscle atrophy in other species.57 In a small group of horses, pergolide treatment was associated with improvement in myofiber-type composition, although myofiber ratio and cross-sectional area were no different. Significant improvement in muscle mass was also observed in a large group of horses with PPID after 6 months of treatment with pergolide mesylate.39 Despite weight loss and muscle mass atrophy, horses with PPID often have abnormal accumulations of fat, most notably in the crest of the neck, tailbase, sheath, and superorbital fossa. This fat accumulation typically predates the weight loss and has a similar pattern as that observed in horses with equine metabolic syndrome (EMS). The similarity of the clinical signs and at-risk breeds of these two diseases has resulted in a misdiagnosis of PPID in animals that have EMS. Unlike what was previously postulated, these two conditions, PPID and EMS, are no longer considered mutually exclusive conditions. There has also been speculation that animals with EMS or sustained obesity with hyperinsulinemia may be at greater risk for developing PPID as they age. Although epidemiologic data are currently lacking to support this association, client-provided anecdotal data suggest this may warrant more critical assessment. Polydipsia and polyuria (PU/PD) occurs in some horses with PPID and is typically mild in severity.44–48,58–59 The mechanism responsible for PU/PD may include (1) compression of the pars nervosa resulting in decreased arginine vasopressin (antidiuretic hormone) production, (2) osmotic diuresis secondary to hyperglycemia and glucosuria, or (3) cortisol induced. Cortisol is thought to cause PU/PD in other species by interfering with the secretion and/or action of arginine vasopression.60 Evidence suggests ACTH and cortisol may inhibit the renin-angiotensin-aldosterone axis as well. The mechanism of PU/PD in horses with PPID has not been extensively examined. Glucosuria is not a common finding in horses with PPID. In one study, two PPID horses with marked hyperglycemia were found to have similar water consumption to normal horses, suggesting that osmotic diuresis is unlikely a major mechanism of PU/PD in the PPID horse.48 Horses with PPID have been reported to be more susceptible to infection, including endoparasitism, bacterial sinusitis, skin infections, abscesses, and respiratory infections.44–45,58–59 The morbidity and mortality of secondary infection in equine PPID has been reported to range from 27% to 48%.46–47,58–59 In the absence of parasite control, a heavy parasite burden is common.61 Routine, quantitative fecal egg counts are recommended to ensure an adequate anthelmintic program. Vigilance on the part of the owner and veterinarian is important in both prevention and early recognition of infections because they may be clinically insidious. Bronchopneumonia was found at necropsy in 7 of 19 horses with PPID.62 Bronchopneumonia should be considered in the PPID horse with fever or tachypnea and ruled out in horses with intermittent hyperhidrosis. Equine PPID is both common and life threatening; therefore early and accurate diagnosis with appropriate intervention is considered critical. Antemortem diagnosis of PPID currently relies on testing hypothalamic-pituitary-adrenal axis responsiveness or measurement of endogenous plasma concentrations of POMC-derived peptides such as ACTH. These tests have been the subject of recent evaluation, and the search for improved testing strategies is ongoing. Accepted best practices for the diagnosis of PPID have evolved significantly over the past 5 years, and further changes are likely to follow. Currently, three antemortem diagnostic tests are most commonly used in practice settings: (1) the overnight dexamethasone suppression test (DST), (2) measurement of endogenous plasma ACTH concentration, and (3) ACTH release following thyrotropin-releasing hormone stimulation. The overnight DST was long considered the “gold standard” method of antemortem PPID diagnosis.63 In the unaffected horse, intramuscular (IM) administration of dexamethasone decreases release of ACTH from the pars distalis, resulting in a serum cortisol concentration of less than 1 µg/dL (27.59 nmol/L) 19 hours after dexamethasone (Fig. 41-7).63 Horses with PPID fail to suppress serum cortisol concentration due to ACTH production from the pars intermedia. Although originally this test was reported to have a sensitivity and specificity of 100%, horses included in that study were selected for having end-stage PPID, resulting in a favorable bias when assessing test performance.64 Although highly specific for PPID, the DST has shown poor performance in identifying horses with earlier PPID. In fact, the DST was only useful in identifying horses with end-stage disease (defined as those with hypertrichosis and grossly enlarged pituitary adenomas) when a random population was assessed using necropsy as the gold standard.64 Measurement of endogenous concentrations of POMC-derived peptides is also useful in the diagnosis of PPID.65 Measurement of ACTH is perhaps the most commonly used method for diagnosis of PPID in ambulatory practice because it requires collection of only a single plasma sample and poses no risk to the patient. Difficulty in sample handling was long cited as a limitation of this test; however, more critical evaluation has found equine ACTH to be more stable in plasma than originally assumed, and as long as a plasma sample remains cooled, separation of plasma can be delayed as long as 12 hours without affecting results.37,47 Measurement of plasma concentrations of ACTH was originally reported to have a sensitivity and specificity of approximately 80% to 90%.47,65 However, evaluation of a larger, randomized equine population has found the test to have a sensitivity and specificity of approximately 70% and 80%, respectively.64 α-MSH concentration, although used in research to assess pars intermedia function, is not commercially available at this time. However, in multiple studies α-MSH concentration did not perform significantly better than ACTH concentration in identification of PPID.26,64–66 A positive test result in an animal with clinical signs of PPID is considered strong diagnostic evidence. However, due to the poor performance of the DST and ACTH concentration at identifying early disease, it is not uncommon for a clinician to have a suspicion of PPID in a patient that has a negative diagnostic test result. When this occurs, further testing using a provocative test is indicated. Presently, the dynamic test that has been shown to most accurately identify PPID is the thyrotropin-releasing hormone (TRH) stimulation test with measurement of plasma ACTH concentration.67–69 The TRH stimulation test was historically recommended for use in horses with a history of laminitis. The diagnosis of PPID was made by demonstrating an increase in serum cortisol concentration 30 to 90 minutes after TRH administration.70 However, further work showed this assay to be unreliable, with false-positive tests common.6 Recently, Beech and colleagues have shown TRH-stimulated ACTH release to be a superior diagnostic test.67–69 TRH is known to be a physiologic releasing factor of the equine pars intermedia.6 Measurement of plasma ACTH, rather than serum cortisol, is a more direct assessment of pars intermedia response, eliminating the influence of adrenal gland response or ACTH bioactivity. It has been demonstrated that in PPID immunoreactive plasma ACTH is not as bioactive as plasma ACTH from the healthy horse.71–72 Therefore in horses with PPID the TRH response may be attenuated at the level of cortisol release. Other strategies to improve the ability to diagnose PPID include combining multiple tests, such as the DST, TRH stimulation test, or plasma ACTH measurement, repeating the same or a different test several months later or testing during the fall (see later) using seasonally specific reference ranges. Seasonal variation in both static and dynamic testing of pars intermedia function has been well documented.22–27,73–74 Testing performed in autumn, between August and October in the northern hemisphere, yields a significantly greater hormone response or concentration than testing performed between November and July.22–27,73–74 Initially, these findings led to the recommendation to avoid endocrine testing in autumn. However, further work has demonstrated that the difference in ACTH concentration between normal horses and those with PPID is much greater during autumn.25 As a consequence, provided seasonally specific reference intervals are available, testing in autumn is more discriminating and therefore preferable to testing during other times of the year. Although unlikely to be practical in most cases, advanced imaging of the pituitary using computed tomography (CT) or magnetic resonance (MR) has also been examined in a limited number of cases as a method for documenting pituitary or pars intermedia enlargement.75–77 Accuracy of CT at estimating width, height, length, and volume of the equine pituitary gland in disarticulated heads from 25 normal horses was determined to be 81% to 93%, 58% to 71%, 88% to 99%, and 43% to 53%, respectively.76 In a more recent study in which CT measurements were performed in a small number of live horses, accuracy was between 92% and 105%, likely due to the larger dose of contrast agent administered.77 Despite the ability to characterize overall size of the pituitary, CT does not provide information on the internal structure of the gland, including the relative size of the pituitary lobes and the presence of microadenomas. MR imaging is the preferred diagnostic imaging modality for pituitary masses in humans, and its usefulness in characterizing the pituitaries of horses is currently being evaluated.78 Postmortem examination of the horse with PPID reveals a grossly enlarged pituitary due to hypertrophy and hyperplasia of the pars intermedia.79–81 The normal horse’s pituitary typically weighs between 1 and 3 g; the affected horse’s pituitary may be two to five times this size. Enlargement may be due to an adenoma (>1 cm), which often contains areas of hemorrhage and necrosis. Alternatively, microadenomatous (≤1 cm) hyperplasia may be present. Melanotropes in the affected gland are pleiomorphic (polyhedral or spindle shaped) with eosinophilic, granular cytoplasm.79–81 Cells are organized into nodules, rosettes, bundles, or follicular structures separated by fine septal tissue. Pigment deposition is common in the pars nervosa, and hemosiderin may be observed when hemorrhage is present. Other lesions include compression of the pars distalis, pars nervosa, or in the case of large tumors that outgrow the sella turcica, compression of the optic chiasm or hypothalamus. Associated lesions of other organs are also common, including those related to disease complications such as laminitis, intestinal parasitism, pneumonia, or sinusitis.62 Mild enlargement of the pars intermedia is also observed in the healthy aged horse,82 and moderate enlargement with microadenomas has been observed in clinically normal horses during the autumn.81 Treatment of PPID is aimed at improving general health and reducing the risk of disease complications such as laminitis and infections. Management practices should be optimized for care of an aged horse. Diet and feeding practices, dental and hoof care, and deworming schedule should be assessed. Feeding of a pelleted diet designed for senior horses along with strategic deworming and correction of dental abnormalities is useful in maintaining the animal’s weight and improving overall general health. Body clipping the horse that fails to shed during warm weather is critical to limit excessive sweating and avoid hyperthermia. Vigilant observation for evidence of infection or laminitis followed by early intervention is important to avoid protracted illnesses. Pharmaceutical interventions for PPID function by decreasing the concentration of circulating POMC peptides and/or cortisol, which theoretically should reduce the risk of disease complications beyond what can be achieved by management alone. Ideally, treatment should also reverse or retard the hyperplastic growth of the pars intermedia, thereby limiting compression of adjacent tissues. The preferred drug for the treatment of PPID is pergolide, a dopamine agonist, which has been shown to improve clinical signs and diagnostic test results in treated animals.36–39,44 An initial dose of 0.002 mg/kg orally every 24 hours has been recommended.44 Response to drug should be assessed by both clinical improvement and normalization of diagnostic test results. If no response is observed in 4 weeks, the dose is increased by 0.002-mg/kg increments monthly until clinical signs and biochemical abnormalities normalize.44 Complications associated with pergolide use in the horse include anorexia, colic, and diarrhea. These are typically dose dependent and often resolve spontaneously following a reduction in dose. Once an effective dose is established, endocrine testing should be repeated every 6 to 12 months to ensure hormonal control is maintained because many horses will require periodic increases in dose over time. Cyproheptidine, a drug with antiserotoninergic, antihistaminergic, and anticholinergic activity, was one of the original drugs used to treat horses with PPID. However, several studies using objectively measurable outcomes failed to show consistent efficacy of the drug.36–37 In addition, although historically inexpensive, the cost of cyproheptidine has increased significantly, making pergolide the more rational treatment choice. Cyproheptidine may be useful as an adjunct therapy in horses resistant to pergolide monotherapy. Cyproheptidine may be added at 0.3 to 0.5 mg/kg orally once daily to horses that show minimal response to 0.006 mg/kg or higher dose of pergolide.44 The prognosis of horses with PPID is not well documented. Many horses live for years following diagnosis, particularly if receiving optimized management. Anecdotal reports of PPID horses that have responded to pergolide for more than 5 years suggest long-term effective treatment is achievable. As with all diseases, early recognition, appropriate intervention, and avoidance of complications are the keys to a positive outcome. Kelsey A. Hart As in other species, in the horse the paired adrenal glands lie immediately craniomedial to the kidneys. Equine adrenal glands weigh approximately 15 g each and are approximately 8 to 10 cm in length, 3 to 4 cm wide, and 1.5 cm thick, though there is much variation in adrenal size and shape among individual horses. Each gland is divided into an outer cortex that secretes corticosteroids and an inner medulla that secretes catecholamines. The adrenal medulla functions as a neuroendocrine organ and is essentially a modified sympathetic ganglion. Preganglionic sympathetic neurons extend from the CNS and synapse directly on adrenal medullary chromaffin cells; when stimulated, these cells release catecholamines directly in the systemic circulation to produce a global sympathetic response. In the horse, the primary adrenal medullary catecholamine appears to be epinephrine, though norepinephrine and dopamine are also produced.1 Diseases affecting the adrenal medulla are rare in horses—as in all species—though adrenal medullary neoplasms are reported. The adrenal cortices are divided into three cellular zones: (1) the outer zona glomerulosa; (2) the middle zona fasiculata, which is the largest zone and comprises approximately 75% of the weight of the gland; and (3) the narrow inner zona reticularis. Cells in the zona glomerulosa are primarily responsible for the secretion of mineralocorticoids (e.g., aldosterone) in response to hypotension and electrolyte derangements (hyperkalemia, hyponatremia). Zona fasiculata cells synthesize and secrete glucocorticoids (e.g., cortisol, corticosterone, 11-deoxycortisol) in response to stimulation by adrenocorticotropic hormone (ACTH) after activation of the hypothalamic-pituitary-adrenal (HPA) axis (Fig. 41-8) by physiologic or pathophysiologic stressors. The predominant circulating glucocorticoid in the horse is cortisol.2 Cells in the zona reticularis also secrete small amounts of glucocorticoids but primarily produce adrenal androgens such as dehydroepiandrosterone (DHEA) and androstenedione. All corticosteroid hormones are structurally similar with a common sterol backbone and one 5-carbon and three 6-carbon rings, and they share a common synthetic pathway originating from cholesterol (see Fig. 41-8). The specific corticosteroids produced by a particular adrenocortical cell depend on which biosynthetic enzymes are expressed in that cell (see Fig. 41-8), which varies among the three functional zones. All steroid hormones are lipophilic and thus are transported in the plasma predominantly bound to plasma proteins, including albumin and steroid-binding globulins such as cortisol-binding globulin (CBG). However, because steroid hormone receptors are located in the cytoplasm of steroid-responsive cells, it is only the free, unbound portion of circulating steroid hormones that is available to enter cells via diffusion across the plasma membrane to bind these intracellular receptors. Binding of the steroid hormone to the receptor causes conformational changes that allow dissociation of regulatory heat shock proteins, permitting the hormone-receptor complex to localize to the nucleus, bind DNA at hormone response elements (HREs), and upregulate or downregulate transcription of steroid hormone–responsive genes. Glucocorticoids are the most abundant circulating adrenal corticosteroid hormone and play an integral role in the endocrine response to stress. The HPA axis (Fig. 41-9) is activated when physiologic, pathophysiologic, or environmental stressors activate peripheral and CNS components, whose signals are then interpreted and integrated in the hypothalamus. This stimulates hypothalamic paraventricular nuclei, resulting in the release of corticotropin-releasing hormone (CRH) into the hypothalamic-hypophyseal portal vessels. CRH then acts locally in the adjacent anterior pituitary gland to stimulate CRH receptors on the cell surface of pituitary corticotroph cells, resulting in the release of ACTH (corticotropin) into the systemic circulation. ACTH binds cell surface receptors (melanocortin2 receptor, MC2R) on adrenocortical cells and stimulates the adrenal glands to synthesize and secrete cortisol. MC2R is a G protein–coupled transmembrane receptor that acts via adenylate cyclase to increase cyclic AMP levels, which then activate biosynthetic enzymes necessary for cortisol synthesis, including 3-β-hydroxysteroid dehydrogenase (3-β-HSD), 17-α-hydroxylase, 21-α-hydroxylase, and 11-β-hydroxylase (see Fig. 41-9). This latter enzyme, present only in glucocorticoid-producing cells, catalyzes the final step in cortisol synthesis from 11-deoxycortisol.3 Cortisol is not stored in adrenocortical cells, but rather is secreted into the systemic circulation immediately following ACTH-induced synthesis. In most adult mammals, including horses, approximately 90% to 95% of circulating cortisol is bound to CBG and albumin.4–8 Many cell types are sensitive to glucocorticoids, permitting cortisol to exert diverse effects necessary for maintenance of homeostasis and stress responses in both health and disease. Essential glucocorticoid-mediated physiologic responses include maintenance of blood pressure, provision of energy to tissues, and control of an appropriate inflammatory response. The sum of cortisol’s systemic effects serves to reduce the physiologic stress that initially activated the HPA axis, to ultimately restore basal HPA axis tone. In addition, cortisol itself acts via negative feedback mechanisms at hypothalamic, pituitary, and adrenal levels to further downregulate HPA axis activity. Thus with an intact HPA axis, plasma cortisol concentrations are maintained at a level appropriate for the existing degree of physiologic stress. Cortisol secretion patterns in adult horses exhibit both ultradian (pulsatile) and circadian rhythms with peak secretion in the morning and the nadir in the evening, similar to many other species.9–12 These ultradian and circadian rhythms are easily disrupted in horses by simple routine changes and illness.9,13 Cortisol responses to ACTH stimulation testing in adult horses are described and suggest adult horses show a comparable dose-dependent effect of exogenous ACTH on cortisol concentrations as is described in other species.11,14–16 In general, an approximately twofold to fivefold increase in cortisol concentration is observed within 30 to 90 minutes after intravenous (IV) administration of a 0.1- to 10-µg/kg dose of exogenous ACTH (cosyntropin) in healthy adult horses14,16; the magnitude of cortisol increase varies with the ACTH dose administered. HPA axis function in the foal differs from both adult horses and other species in several key ways. Compelling evidence shows that maturation of the HPA axis occurs in the days just prior to parturition and continues during the first several weeks of life, much later than is described in other species.17–20 Premature foals have lower serum cortisol and higher ACTH concentrations than full-term foals immediately postpartum.20 These low baseline cortisol and concurrent high ACTH concentrations in premature foals imply that foals may have impaired adrenocortical sensitivity to ACTH, limited cortisol synthetic capacity, or both, as compared with adult horses. Data from ACTH stimulation test responses in foals support this theory. Premature foals show a blunted cortisol response to exogenous ACTH, with only a 28% increase in plasma cortisol 30 to 60 minutes following stimulation, as compared with a 208% increase in normal term foals.17 Evidence suggests that the fetal foal’s adrenal gland may be incapable of synthesizing cortisol and some other steroid hormones until late in gestation. Fetal foals do not express substantial levels of the key steroidogenic enzymes cholesterol side chain cleavage enzyme, 17α-hydroxylase, and 3-β-HSD until just before parturition in the foal,21,22 much later than is described in other species.23 Furthermore, adrenocortical function may not be fully mature at birth even in full-term foals. Though increased at birth due to the stress of parturition, by 12 to 24 hours of age, mean basal cortisol concentrations are onefold to twofold lower in healthy neonatal foals than reported mean concentrations in healthy adult horses despite comparable or higher concurrent ACTH concentrations in foals.24–28 This apparent decreased cortisol response to endogenous ACTH in the full-term neonatal foal is supported by further evidence of limited cortisol responses to exogenous ACTH. Insulin-induced cortisol responses in foals within 12 hours of birth are less than half of the responses achieved in 7- to 14-day-old foals, 29 and healthy term neonatal foals aged 1 to 7 days are reported to have approximately half the magnitude of cortisol response to ACTH stimulation testing than adult horses.14,25 This decreased adrenocortical responsiveness appears to persist during the first few months of life, as 12-week-old foals show significantly greater cortisol responses to a low-dose (0.1 µg/kg) ACTH stimulation test than younger foals.30 Finally, foals differ from adult horses in their plasma cortisol binding capacity and cortisol secretion and metabolism patterns. In foals younger than 1 week of age, 30% to 60% of circulating cortisol is free, in contrast to 5% to 10% in adult horses.31 This is likely due to decreased CBG concentrations in foals—as has been shown in infants32,33—though an assay for equine CBG is not currently available. Free cortisol is preferentially excreted and metabolized over bound cortisol,34 so it is not surprising that increased cortisol clearance rates in foals as compared with adult horses have recently been described.12 Healthy full-term foals appear to be able to increase their daily cortisol production rate to compensate for this increased clearance, though as serum total cortisol concentration at 1 week of age remains significantly lower in foals than adult horses,12 foals’ capacity to further and appropriately increase cortisol production in severe stress may be limited. It is likely that impaired cortisol responses in neonatal foals result from multiple factors, including decreased cortisol synthetic capacity and increased cortisol clearance and possibly other as yet undefined differences in ACTH sensitivity or activity. Regardless of the cause, this evidence of fetal and neonatal HPA axis immaturity is certain to substantially affect foals’ stress responses during the neonatal period. Mineralocorticoids (e.g., aldosterone) are produced in small quantities relative to glucocorticoids but have equally important physiologic roles. Angiotensin II, via activation of the renin-angiotensin-aldosterone system by hypovolemia/hypotension or increases in plasma osmolality, and increased plasma potassium concentration are the primary stimulants of aldosterone synthesis and secretion. ACTH is also necessary for aldosterone synthesis, but ACTH administration to healthy adult horses resulted in a decrease in plasma aldosterone concentration in contrast to increases seen in other species.35,36 Like glucocorticoids, aldosterone circulates in the plasma bound to albumin or steroid-binding globulins and primarily acts by binding cytosolic mineralocorticoid receptors to alter transcription of genes necessary for sodium and potassium transport. Mineralocorticoid receptors are predominantly expressed in sodium-transporting epithelia in the distal renal tubules and colon, with lesser expression in the rest of the intestinal tract and the heart. Aldosterone induces transcription of an aldosterone-regulated kinase that increases activity of apical membrane sodium channels.37 The net effect of this is increased sodium flux across epithelial cells, resulting in increased renal and intestinal sodium and water resorption and concurrent stimulation of potassium excretion via the basolateral Na+/K+ ATP-ase.37 Mineralocorticoids are vital for appropriate fluid and electrolyte balance, and mineralocorticoid deficiency rapidly results in hyponatremia, hyperkalemia, and hypovolemia. Reported resting and exercising plasma concentrations of aldosterone in horses have been described.38,39 In contrast to their limited cortisol responses to appropriate stimuli, newborn foals appear to be able to mount a substantial aldosterone response to hypovolemia/hyponatremia.40,41 In fact, one study suggested that the aldosterone response in foals appears to be exaggerated in comparison to that in adult horses and may reflect differences in tubular sensitivity to aldosterone between neonates and adults.41 However, well-established reference ranges for plasma aldosterone concentrations in foals are not currently available. In adult males, circulating androgens are primarily of testicular origin, but in females more than half of plasma androgens may originate from the adrenal cortices. ACTH is necessary for the synthesis and secretion of adrenal androgens, but another factor (or factors) that is not yet identified is also required for adrenal androgen synthesis.11 Adrenal androgens are also transported in the plasma bound to albumin and CBG and act via cytosolic steroid hormone receptors to modulate gene transcription. DHEA and androstenedione can be extraglandularly metabolized to active testosterone and estrogens and play a particularly important role in early pubertal sexual development in most species. Androstenedione concentrations in adult horses are reported, and sex and age variations in adrenal androgen concentrations may be relevant but at present are poorly understood in horses and foals.27,42 Increased adrenal androgen production is postulated to play a role in mares with estrus-related behavior problems, though this has not been definitively proven.27 Critical illness–related corticosteroid insufficiency (CIRCI) has been recently described in septic foals and adult horses with colic. CIRCI (also known as relative adrenal insufficiency) is best understood in people and is defined as an insufficient cortisol response or inadequate cortisol activity for the existing degree of critical illness.43–47 Although serum cortisol concentrations in patients with CIRCI are often increased relative to resting concentrations in healthy individuals, the cortisol response remains insufficient for the markedly increased demands of acute severe illness.43,48–50 In people CIRCI is reported to occur secondary to severe illness, such as sepsis or acute respiratory distress syndrome (ARDS), severe trauma, or major surgery, and appears to affect up to 40% to 60% of adult and pediatric patients with sepsis and septic shock.45,46,48 In CIRCI, the cortisol insufficiency is transient and resolves if the patient survives the primary illness. However, due to the vital role the HPA axis plays in the physiologic response to the stress of illness, the occurrence of CIRCI during critical illness substantially worsens the morbidity and mortality of the primary disease. For instance, septic people with CIRCI have significantly higher incidence of multiple organ failure and decreased survival rates as compared with septic patients with appropriate HPA axis function.44–46,49,51,52 Current understanding of HPA axis function in critically ill horses and foals is limited; to date, CIRCI is best characterized in septic neonatal foals.53–58 Although basal ACTH and cortisol concentrations are higher in septic foals than healthy age-matched control foals and in nonsurviving foals (as one would expect with the stress of illness), several studies have reported significantly increased ACTH-to-cortisol ratios in nonsurviving septic foals.53,55,57 Such high ACTH-to-cortisol ratios, with high ACTH concentrations and low corresponding cortisol concentrations, suggest cortisol synthesis failure at the level of the adrenal gland may occur in the septic full-term foal. Findings from ACTH stimulation tests in hospitalized foals also provide evidence for adrenocortical dysfunction in this population. Peak and delta cortisol (peak–basal cortisol concentration) responses to ACTH stimulation tests do not appear to differ between healthy and ill foals, but increased disease severity and poorer prognoses were associated with decreased cortisol responses to ACTH stimulation in hospitalized foals.56,58 Specifically, nonsurviving foals had significantly lower delta cortisol responses to low-dose ACTH stimulation as compared with survivors,58 and foals that met criteria for CIRCI (as characterized by an inadequate delta cortisol response to a high-dose ACTH stimulation test) had a significantly greater incidence of shock, multiple organ dysfunction syndrome, and nonsurvival than foals with an adequate cortisol response to ACTH.56 When human diagnostic criteria for CIRCI59 based on inadequate delta cortisol responses to high-dose ACTH stimulation were adapted and applied to hospitalized foals 1 week of age and younger, approximately 50% of all hospitalized foals and a subgroup of septic foals met these criteria.56 The incidence of CIRCI in critically ill horses has only been examined in one study to date, but findings suggest it may also occur in this population.60 Twenty-four percent of severely ill horses had inappropriately low basal cortisol concentrations at hospital admission, and 85% of these horses had inadequate delta cortisol responses to ACTH stimulation.60 Marked adrenal hemorrhage at necropsy was also seen in nonsurviving horses in this study.60 In concert, this evidence suggests that CIRCI occurs in critically ill and septic neonatal foals and in critically ill horses with comparable frequency and impact as in people with similar illnesses. The specific pathophysiologic mechanisms leading to the development of CIRCI are poorly understood in all species and have not yet been systematically evaluated in horses or foals. A combination of factors is likely involved in the development of HPA axis dysfunction in CIRCI, including direct damage to HPA axis components from the primary disease, inhibition of cortisol production by medications used to treat the primary disease, and suppression of activity of one or more components of the HPA axis by infectious organisms or the patient’s own immune and inflammatory response.44,45 Periods of hypotension associated with hypovolemic, endotoxic, or septic shock can result in decreased adrenal perfusion and ischemic injury to the metabolically active adrenocortical cells. If this occurs for prolonged periods or to an extreme degree, the damage may be irreversible, resulting in severe adrenocortical hemorrhage and necrosis known as Waterhouse-Friderichsen syndrome in people61; similar adrenal pathology has been recently described in critically ill horses and foals.60,62 Milder adrenal circulatory insults may result in transient injury and dysfunction. In addition, drugs like the anesthetic agent etomidate and several antimicrobial agents (e.g., ketoconazole, rifampin) can directly inhibit adrenocortical cortisol synthesis.45,63 It is the patient’s own immune and inflammatory response, though, that appears to play the most vital role in the development of CIRCI. Bacterial components such as endotoxin, as well as host proinflammatory cytokines, all play an appropriate and important role in initiating and maintaining the HPA axis response to critical illness by directly stimulating HPA axis activity at multiple levels.44,61,64–66 However, bacterial ligands and inflammatory cytokines may also be capable of suppressing HPA axis function at one or more levels, in the face of an overwhelming bacterial infection or excessive host inflammatory response. For example, inducible nitric oxide synthase-mediated death of hypothalamic neurons in cardioregulatory centers has been described in patients that died of septic shock and may play a role in HPA axis dysfunction in such patients.67 Bacterial endotoxin has been shown to directly decrease pituitary CRH receptor gene expression in both rats and cattle.68,69 In addition, the proinflammatory cytokine TNF-α, which is increased in sepsis, can directly impair both pituitary ACTH release and adrenocortical cortisol synthesis.45 Finally, cholesterol availability for corticosteroid synthesis may be limited during sepsis, as several studies have shown decreased levels of plasma high-density lipoprotein (HDL) in critically ill individuals and correlated these decreased HDL levels with blunted cortisol responses to ACTH stimulation testing.45 Interactions between the adrenal axis and the immune response in horses are not well characterized, but equine adrenocortical tissue has been shown to directly secrete IL-6, IL-10, and TNF-α in an ex vivo model,70 and a positive association between plasma ACTH concentration and leukocyte IL-6 expression has recently been shown in septic foals.71 Several studies suggest that peripheral tissue cortisol resistance may develop in some critically ill patients. Specifically, impaired GR binding affinity has been documented in an ovine model of acute lung injury72 and a rodent burn model,73 where it was partially ameliorated when TNF-α and IL-1β were reduced by neutralizing antibodies. Furthermore, nuclear localization of cortisol-GR complexes was impaired in human leukocyte cells exposed to plasma from patients that died of acute respiratory distress syndrome.74 Thus the complex relationship between endocrine and immune regulation of HPA axis function at multiple levels determines overall cortisol production and activity during sepsis. Any imbalance in these interactions can result in absolute or functional cortisol insufficiency and may play a critical role in the pathogenesis of CIRCI; further study of specific mechanisms resulting in CIRCI in horses and foals is needed.
Endocrine and Metabolic Diseases

Pituitary and Hypothalamus
Physiology of the Equine Hypothalamic-Pituitary Axis
Equine Pituitary Pars Intermedia Dysfunction
Pathophysiology
Clinical Signs
Diagnosis of Pituitary Pars Intermedia Dysfunction
Necropsy Findings
Treatment and Prognosis
Adrenal Glands
Critical Illness–Related Corticosteroid Insufficiency
Definition and Epidemiology
Pathophysiology
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree