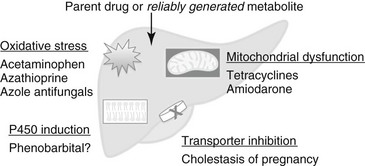
Chapter 140 An R value greater than 5 indicates hepatocellular injury, an R less than 2 indicates cholestatic injury, and an R of 2 to 5 represents a mixed pattern. For dose-dependent, or intrinsic, hepatotoxicity, toxicity increases with increasing dose in one or more species, and virtually all members of a population or species are affected at high enough doses. Dose-dependent hepatotoxicity may be caused by the parent drug or by a metabolite generated reliably in the treated species (Figure 140-1). These reactions are relatively predictable, and therapeutic drug monitoring may be helpful in prevention. They require a dose reduction but usually not permanent drug discontinuation. Figure 140-1 Dose-dependent, drug-associated hepatotoxicity typically is caused by either the parent drug or a consistently generated metabolite. Drugs can inhibit transporter pumps and lead to a functional cholestasis; this occurs with endogenous hormone metabolites during pregnancy. Many drugs yield reactive metabolites that cause oxidative stress; examples include acetaminophen, azathioprine, and azole antifungals. For these compounds, antioxidant supplementation may be effective. Drugs that interfere with mitochondrial function can lead to steatosis from inhibition of fatty acid β-oxidation or may lead to more severe hepatocellular damage because of impaired cellular respiration. Finally, drugs that act as P450 inducers, such as phenobarbital, may mediate hepatotoxicity by chronic bioactivation of environmental toxins. Hepatotoxicity from phenobarbital can range from subclinical increases in serum bile acids to clinical hepatopathy to fulminant liver failure (Dayrell-Hart et al, 1991). Signs typically develop after a year or more of phenobarbital treatment, and prolonged duration of administration is associated with degree of histologic injury in epileptic dogs. Typical histologic findings in dogs with clinical signs are bridging portal fibrosis, bile duct hyperplasia, and nodular regeneration. Dogs with phenobarbital hepatotoxicity improve clinically after phenobarbital dose reduction (Dayrell-Hart et al, 1991). However, liver enzyme abnormalities can develop in phenobarbital-treated dogs without histologic liver injury, and higher phenobarbital dosages and serum drug concentrations have not been correlated with the development of abnormal serum bile acids in epileptic dogs. These findings suggest that phenobarbital hepatotoxicity is dose dependent, with modifying factors that are not understood. Ketoconazole, itraconazole, and fluconazole can lead to increases in serum ALT in dogs, although clinical signs such as jaundice are uncommon. Mild, clinically insignificant increases in ALT also have been reported in cats treated with itraconazole and fluconazole. In dogs with blastomycosis, higher dosages of itraconazole (10 mg/kg/day) were associated with a greater risk of ALT abnormalities than 5 mg/kg/day, with no difference in efficacy (Legendre et al, 1996). Further, increases in ALT and ALP were correlated with itraconazole plasma concentrations, which supports a dose-dependent mechanism. Amiodarone leads to clinically significant hepatotoxicity in about 45% of dogs treated for refractory atrial fibrillation and ventricular arrhythmias, a median of 16 weeks after starting maintenance therapy (Jacobs et al, 2000; Kraus et al, 2009). Predominant increases in ALT typically are observed, with or without hyperbilirubinemia and neutropenia. These abnormalities slowly resolve over 1 to 3 months after drug discontinuation. Toxicity in animal models has been attributed to two oxidative metabolites, mono-N-desethylamiodarone (MDEA) and di-N-desethylamiodarone (DDEA), which generate reactive oxygen species that uncouple oxidative phosphorylation and lead to mitochondrial damage.
Drug-Associated Liver Disease*
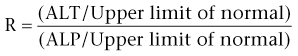
Dose-Dependent Hepatotoxic Drugs
Phenobarbital
Azole Antifungals
Amiodarone
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


