Chapter 4 Disorders of Chloride
Hyperchloremia and Hypochloremia
“Whereas for a long time it was assumed that chloride ions were reabsorbed entirely passively with sodium — the “mendicant” role of chloride — more recent studies suggest that several distinctive reabsorptive transport mechanisms operate in parallel.”86
Chloride is present in plasma at a mean concentration of approximately 110 mEq/L in dogs and 120 mEq/L in cats.17 Chloride concentration in venous samples is 3 to 4 mEq/L lower than those in arterial samples when cells are separated from plasma anaerobically.92 The intracellular concentration of chloride is much lower than its plasma concentration and is dependent on the resting membrane potential of the cell. Muscle cells, for example, have a resting membrane potential of approximately −68 mV and an average chloride concentration ([Cl−]) of 2 to 4 mEq/L, whereas red blood cells have a resting membrane potential of approximately −15 mV and an average [Cl−] of 60 mEq/L.55 This higher intracellular concentration of chloride ions in erythrocytes allows chloride to move in and out of the red blood cells very effectively, as dictated by electrical charges on either side of the cell membrane. This is an important difference from other cells and is the basis of the so-called “chloride-shift” in the red cell membrane.55 The chloride ion distribution in various body fluids is summarized in Box 4-1.
Box 4-1 Chloride Ion in Various Body Fluids
From de Morais HSA: Chloride ion in small animal practice: the forgotten ion, J Vet Emerg Crit Care 2:11–24, 1992.
Chloride metabolism
Gastrointestinal tract
Under normal conditions, humans produce 1 to 2 L of gastric juice daily. The sodium concentration ([Na+]) and [Cl−] of gastric juice is quite variable, ranging from 20 to 100 mEq/L and 120 to 160 mEq/L, respectively.76 In the jejunum, sodium is absorbed actively against small electrochemical gradients and also through relatively large mucosal “pores” in the proximal bowel. Chloride absorption in the jejunum generally follows sodium to maintain electroneutrality. It is believed that chloride reabsorption in the jejunum occurs via the paracellular route in response to the transepithelial potential generated by active sodium transport.24 The ileum is less permeable to ions than the jejunum. Absorption of chloride and secretion of bicarbonate in the ileum are coupled by processes that may involve active transport of one or both ions. Highly efficient absorption of sodium and chloride occurs in the colon, where 90% of the sodium and chloride entering is reabsorbed. There appears to be no direct or indirect coupling between sodium and chloride or bicarbonate reabsorption in the distal colon. Active chloride reabsorption and bicarbonate secretion occur in the distal colon. Chloride also can be secreted in the jejunum, ileum, and colon.24,76 Pancreatic juice usually is not rich in chloride ions. However, there is a reciprocal relationship between chloride and bicarbonate concentration in pancreatic fluid that is dependent on flow rate, with chloride being the major anion at lower rates of secretion.24
Kidneys
The kidneys play an important role in the regulation of plasma chloride concentration. After sodium, chloride is the most prevalent ion in the glomerular ultrafiltrate. Most of the chloride filtered is reabsorbed in the renal tubules. The traditional view of epithelial transport in the kidneys represents the chloride ion as an obedient passive partner that follows the actively transported sodium ion. This view does not apply to many epithelia, including specific nephron segments. Chloride transport is intimately related to sodium and fluid transport and to cellular acid-base metabolism.86
Chloride reabsorption in the proximal tubule is actively and passively linked to active sodium reabsorption. A formate-chloride exchange mechanism exists in the luminal membrane of proximal tubular cells and is responsible for active chloride reabsorption.81 Reabsorbed chloride returns to the systemic circulation at the basolateral membrane primarily by a potassium chloride (K+– Cl−) cotransporter. Of filtered chloride, approximately 50% to 60% is reabsorbed by the proximal convoluted and straight tubules. Chloride reabsorption occurs transcellularly in the thick ascending limb of Henle’s loop, leading to the generation of a lumen-positive transepithelial voltage. Sodium is reabsorbed transcellularly or paracellularly, and the transepithelial voltage drives the latter process. Chloride ion delivery is the rate-limiting step in this process, and net sodium chloride (NaCl) transport increases directly with fluid [Cl−] concentration. Loop diuretics such as furosemide and bumetanide act in the loop of Henle by competing for the chloride site on the Na+-K+-2 Cl−carrier.24,46,81,86
A comprehensive model explaining sodium chloride transport in the distal tubule is not yet available. This is because of, in part, the cellular heterogeneity of this nephron segment and differences among species and because a portion of this nephron segment is not accessible to micropuncture techniques in rats, the most extensively studied species. Thiazide diuretics act by inhibiting the Na+– Cl− carrier in the early distal tubule, apparently at the chloride site.46 Conversely, loop diuretics do not block NaCl reabsorption at this site. Chloride ion transport in the collecting tubule is closely related to bicarbonate transport.81 Little is known about chloride transport in the medullary collecting tubules. In the cortical collecting tubules, however, the paracellular pathway, which is highly conductive for chloride ions, is an important route for reabsorption of chloride by diffusion down an electrochemical gradient. An increase in the lumen-positive transepithelial potential difference (TPD) decreases net chloride reabsorption, whereas a decrease in TPD increases chloride reabsorption. Therefore, hormones that change TPD in the cortical collecting tubule can affect chloride reabsorption. Experimentally, administration of deoxycorticosterone acetate (DOCA) twice daily resulted in a mild increase in [Na+] and no change in [Cl−].54 The resulting increase in strong ion difference (SID; the difference between all strong cations and all strong anions in plasma; see Chapter 13) was associated with a mild increase in bicarbonate ion concentration ([HCO3−]). Administration of DOCA in sodium-supplemented dogs caused a significant increase in plasma [Na+] and [HCO3−] with no change in plasma [Cl−].64 When NaHCO3, instead of NaCl, was added to the diet, [Na+] and [HCO3−] increased significantly, whereas [Cl−] decreased. Increased urinary loss of chloride is believed to be associated with hyperadrenocorticism. In a study of 117 dogs with hyperadrenocorticism, only 12 had [Cl−] below 105 mEq/L.61 However, 25 of these dogs had hypernatremia, and the [Cl−] could have been low relative to the [Na+]. The mean [Na+] was 149.9 mEq/L, and the mean [Cl−] was 108 mEq/L (mean [Cl−] after correcting for changes in free water was 105 mEq/L). The cortical collecting duct is the main site of action for mineralocorticoids and glucocorticoids.19 Administration of DOCA increases TPD in rats and rabbits, increasing sodium reabsorption in the cortical collecting tubules. Such an effect could explain the observed changes in chloride and sodium concentrations in dogs with hyperadrenocorticism.
Chloride and acid-base balance
Metabolic acidosis
Metabolic acidoses are traditionally divided into hyperchloremic (normal anion gap [AG]) and normochloremic (high AG) based on the AG and [Cl−]. The AG is the difference between measured cations (sodium and potassium) and measured anions (chloride and bicarbonate) (see Chapters 9 and 10). Physiologically, there is no AG because electroneutrality must be maintained and the AG is the difference between the unmeasured anions (UA) and unmeasured cations (UC+). The AG is a simplification that is helpful clinically. Metabolic acidosis usually results from an increase in the concentration of a strong anion. Strong anions are anions that are completely dissociated at the pH of body fluids (e.g., Cl−, lactate, ketoanions). If the strong anion added is chloride, the sum of the measured anions ([Cl−] + [HCO3−]) will remain the same, and the AG will not change (so-called hyperchloremic or normal AG acidosis). If the strong anion added is an unmeasured anion (e.g., lactate), [Cl−] will remain normal, whereas [HCO3−] will decrease. The sum of the measured anions will decrease, thus increasing the AG (so-called normochloremic or high AG acidosis).
The acid-base status of plasma is regulated by changing Pco2 in the lungs and SID in the kidneys, the latter being accomplished mainly by differential reabsorption of sodium and chloride ions in the renal tubules. Chloride is the most prevalent strong anion in the ECF. At a constant [Na+], a decrease in [Cl−] increases SID causing hypochloremic alkalosis, whereas an increase in [Cl−] decreases SID causing hyperchloremic acidosis. The effects of increasing [Cl−] without changing [Na+] can be understood when considering a fluid with an SID = 0 (e.g., 0.9% NaCl where [Na+] = [Cl−] and thus SID = [Na+] − [Cl−] = 0).14 It is known that 0.9% NaCl administration leads to metabolic acidosis. The classic explanation is that infusion of a fluid without bicarbonate dilutes [HCO3−] in plasma and leads to acidosis. However, the degree of acidosis after normal saline infusion correlates best with the amount of chloride given and with the increase in serum [Cl−].98 There was a weaker correlation with the volume administered and no increase in plasma volume, calling into question the traditional concept of dilutional acidosis.
Chloride in metabolic alkalosis
Chloride participates in the genesis, maintenance, and correction of metabolic alkalosis because decreases in [Cl−] increase SID, causing metabolic alkalosis. The role of chloride is supported by the inverse relationship between chloride and bicarbonate in metabolic alkalosis,6 the fact that chloride depletion is accompanied by increased plasma [HCO3−],74 and the fact that chronic metabolic alkalosis cannot be produced experimentally if chloride is available in the diet.59 In addition, during recovery from chronic hypercapnia, the compensatory increase in [HCO3−] will not normalize if dietary chloride is restricted.88
Chloride was first linked to metabolic alkalosis in dogs when MacCallum and colleagues63 observed hypochloremia and an increase in “alkali reserve” in dogs with loss of gastric fluid caused by pyloric obstruction. The classic hypothesis associates the genesis and maintenance of metabolic alkalosis primarily with volume contraction. According to this hypothesis, volume depletion accompanying alkalosis augments fluid reabsorption in the proximal tubules. Alkalosis is maintained because bicarbonate ions are preferentially reabsorbed in this segment.40 Volume expansion suppresses fluid and bicarbonate reabsorption, and more bicarbonate and chloride ions are delivered to distal nephron segments, which possess greater capacity to reabsorb chloride than bicarbonate. Chloride then is retained, bicarbonate is excreted, and alkalosis is corrected.40 In addition to volume expansion, provision of chloride was also a feature of studies used to substantiate this hypothesis. The classic hypothesis can be viewed from a different perspective in which changes in chloride are the cause of the alkalosis.39 In rats, chloride ion depletion alone plays a role in the genesis and maintenance of metabolic alkalosis.35–37,62 In rats with chronic hypochloremic alkalosis, chloride repletion (and correction of alkalosis) can be achieved without administration of sodium, without volume expansion, and without an increase in the glomerular filtration rate (GFR).96 The correction phase is associated with a decrease in plasma renin activity but with no change in plasma aldosterone concentration. It also has been shown that maintenance and correction of hypochloremic alkalosis primarily are dependent on total body chloride and its influence on renal function, and not on the demands of sodium and fluid homeostasis.38 Ultimately, the correction of alkalosis is dependent on the kidneys.40 The principal mechanisms by which the kidneys correct metabolic alkalosis probably operate in the collecting ducts, especially in the cortical segment, where HCO3– can either be secreted or reabsorbed.39
Expanding the ECF without providing chloride does not correct hypochloremic alkalosis. Furosemide-induced hypochloremic alkalosis in humans eating an NaCl-free diet supplemented with 60 mEq potassium per day can be corrected with orally administered KCl without changes in weight or ECF volume.84 In this study, five NaCl-depleted control subjects were given furosemide and a combination of KCl and NaCl intravenously to maintain their sodium deficit while correcting their chloride deficit. Subjects who were selectively sodium depleted did not become alkalotic.84 It also has been shown that a 25% increase in ECF volume (created by intravenous infusion of 6% bovine albumin in 5% dextrose) has no effect on hypochloremic alkalosis in a rat model of hypochloremic alkalosis.38
These studies demonstrate that ECF volume, GFR, effective circulating volume, and sodium balance are not independent variables in the generation and maintenance of metabolic alkalosis.72 However, it still could be concluded that chloride induces potassium conservation that in turn inhibits bicarbonate reabsorption, because potassium balance was corrected even in studies during which choline-Cl instead of KCl was used to correct the alkalosis. When NaCl is supplied without potassium, however, alkalosis is corrected despite a persistent potassium deficit,4,71 and administration of potassium without chloride does not correct alkalosis.56 It has been speculated that hypokalemia in rats may cause hypochloremia by impairing recycling of potassium at the luminal membrane in the thick ascending limb of Henle’s loop. This, in turn, impairs the effectiveness of the Na+-K+-2 Cl− carrier, decreasing net chloride reabsorption.40 It still is controversial whether hypokalemia induces metabolic alkalosis in humans.34 Contrary to what occurs in rats, isolated potassium deficiency in dogs causes mild metabolic acidosis.8,9 Chronic potassium depletion also is associated with metabolic acidosis in cats.25
Studies in rats with experimentally induced normovolemic and hypovolemic hypochloremic alkalosis showed no difference in the renal handling of chloride and bicarbonate between alkalotic and normal animals in the proximal convoluted tubule, loop of Henle, or distal convoluted tubule.40 Key adjustments in anion excretion during the maintenance and correction of hypochloremic alkalosis were suspected to occur in the collecting tubule, especially in the cortical segment.39 Sodium-independent chloride and bicarbonate transport, and secretion or reabsorption of HCO3− occur at this site.39,40 Alterations in the delivery of HCO3− and Cl− to the collecting tubules also may be important.39
The chloride depletion hypothesis for the genesis and maintenance of metabolic alkalosis was proposed as an extension of the classic hypothesis.39,40 It states that chloride alone is essential for correction of the hypochloremic alkalosis and that it does so by a renal mechanism. Volume depletion is a common but not essential feature of the maintenance phase of alkalosis, and its persistence does not preclude correction of alkalosis. If adequate chloride is provided, restoration of depleted volume, however, may hasten correction of alkalosis by increasing the GFR and decreasing proximal tubular reabsorption of fluid and bicarbonate.39,40 The manner by which exogenous Cl− repletion is detected and the kidneys are signaled to excrete HCO3−, and the cellular mechanisms by which these events occur in the various nephron segments, remain to be determined.39
Role of chloride in adaptation to acid-base disturbances
Chloride excretion is an important mechanism in the kidneys’ adaptation to metabolic acidosis and chronic respiratory acid-base disturbances. In metabolic acidosis, the kidneys increase net acid excretion (primarily by enhanced NH4Cl excretion) beginning on day 1 and reaching a maximum after 5 to 6 days.81 The increase in chloride ion excretion without an associated increase in sodium ion excretion increases plasma SID and returns [HCO3−] and pH toward normal.
The increase in Pco2 in chronic respiratory acidosis causes intracellular [H+] to increase in the renal tubular cells, resulting in stimulation of net acid excretion (primarily as NH4Cl).81 Chloruresis, negative chloride balance, enhanced fractional and absolute bicarbonate reabsorption, and enhanced net acid excretion typically are associated with the renal response to chronic respiratory acidosis.40 The loss of chloride ions in the urine decreases urinary SID, because Cl− is accompanied by NH4+ (a weak cation) rather than Na+. Thus, plasma SID and consequently [HCO3−] are increased. Hypochloremia is a common finding in dogs with chronic hypercapnia.65,78,87,94 Conversely, renal H+ ion excretion is decreased in chronic respiratory alkalosis. This effect probably is mediated by a decrease in intracellular [H+]. In this setting, there is a decrease in NH4Cl excretion in urine and an increase in renal reabsorption of Cl−. The increase in Cl− reabsorption decreases plasma SID and consequently [HCO3−] is responsible for the hyperchloremia observed in dogs with chronic hypocapnia.42
Clinical approach to chloride disorders
Corrected chloride
Changes in chloride concentration can result from changes in water balance or can be caused by a gain or loss of chloride. When changes in [Cl−] are caused by water balance alterations (i.e., increase or decrease in free water), [Na+] also changes. Changes in [Cl−] and [Na+] are proportional in this setting (e.g., a 10% gain in free water decreases [Cl−] and [Na+] by 10%). To account for changes in water balance, [Cl−] must be evaluated in conjunction with evaluation of changes in [Na+].17 Therefore, patient [Cl−] is “corrected” for changes in [Na+] concentration:17
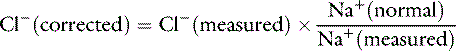
where Cl− (measured) and Na+ (measured) are the patient’s chloride and sodium concentrations, respectively, and Na+ (normal) is the mean normal sodium concentration.
Assuming mean values for [Na+] of 146 mEq/L in dogs and 156 mEq/L in cats, and for [Cl−] of 110 mEq/L in dogs and 120 mEq/L in cats,20 the Cl− (corrected) can be estimated17 in dogs as:
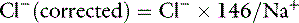
and in cats as:
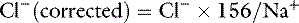
Normal Cl− (corrected) is approximately 107 to 113 mEq/L in dogs and approximately 117 to 123 mEq/L in cats.17 These values may vary between different laboratories and different analyzers. Newer analyzers report higher chloride values unless the chloride calibration is deliberately changed.102 Using the Cl− (corrected) permits the division of chloride disorders into artifactual and corrected chloride changes (Table 4-1). In artifactual chloride changes, changes in free water are solely responsible for the chloride changes, whereas in corrected chloride changes, chloride itself is primarily changed. Algorithms for the evaluation of chloride abnormalities are presented in Figures 4-1 through 4-3.

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


