Robert J. Mackay, David C. Van Metre, Consulting Editors Mary O. Smith • Lisle W. George • David C. Van Metre Cerebrospinal fluid (CSF) is partly derived from and in equilibrium with the extracellular fluid that bathes the brain and spinal cord parenchyma.1–3 The composition is an indicator of the state of the intrathecal contents. CSF is produced by a combination of ultrafiltration of plasma and active secretion.5 The sites of CSF production are the choroid plexuses of the lateral, third, and fourth ventricles; the ependymal lining of the ventricular system; the pia arachnoid; and the meningeal blood vessels. The CSF in the ventricular system flows caudally and diffuses out of the lateral apertures in the fourth ventricle. It then circulates around the brain and spinal cord. Circulation of CSF is achieved through regional pressure changes caused by spinal motion and pulsations of blood vessels. Resorption of CSF occurs from both the cranial cavity (75%) and the spinal canal (25%).6 Some resorption of brain CSF occurs at the arachnoid villi associated with large veins and sinuses, but most resorption occurs at the cribriform plate, into nasal lymphatics.7,8 Resorption of spinal CSF occurs into the lymphatics associated with spinal nerves. The predominant direction of CSF flow in the spinal canal is cranial to caudal. CSF can be collected from the cisterna magna when the site of interest is the brain, whereas it should be collected from the lumbar subarachnoid space when the lesion is in the spinal cord. In large animals, however, the risks associated with the general anesthesia required for cisterna magna tap are considerable, so lumbar puncture is usually the most suitable technique for CSF collection in these species, regardless of the site of the lesion.9,10 For collection of CSF by lumbosacral puncture, the animal is lightly sedated and the skin of the dorsal midline over the junction of the sixth lumbar (L6) and first sacral (S1) vertebrae is surgically prepared (Fig. 35-1). A variety of standard sedative protocols are suitable, although xylazine has been shown to reduce CSF pressure.11 Correct placement of the spinal needle is more easily achieved with the animal standing. The proper anatomic site for insertion of the spinal needle is between the dorsal spinous process of L6 cranially and S1 caudally and the two tuber sacrales laterally. The overlying skin forms a depression that can be recognized by palpation, although this may be difficult in well-muscled horses. The correct site for needle placement can also be located by determining (1) the dorsal midline at the highest point of the quarters or (2) the point where a line drawn between the caudal aspects of the two tuber coxae intersects the midline. The skin is anesthetized with 2% lidocaine, and a 1-cm incision is made with a No. 15 scalpel blade. A 6- to 9-inch, 18- to 20-gauge spinal needle is inserted perpendicularly through the incision and advanced until the tip punctures the lumbosacral cistern (a The spinal needle is advanced gently to the floor of the spinal canal. Passage of the needle through the terminal spinal cord or the cauda equina does not cause subsequent neurologic abnormalities. When the needle is seated in the spinal canal, gentle negative pressure can be applied by withdrawing spinal fluid into a series of 3-mL syringes. If frank blood is obtained, the tip of the needle is probably in one of the ventral vertebral sinuses. The needle should be withdrawn a few millimeters, and a clean syringe should be attached to the hub. Compression of the jugular vein causes engorgement of the ventral vertebral plexus, which increases CSF pressure in the lumbosacral cistern. General anesthesia is required for a cisterna magna tap. After the patient has been anesthetized, the dorsal area of the neck overlying the atlantooccipital joint is surgically prepared. The patient’s head is held flexed at a right angle to the neck, with the sagittal plane of the head parallel to the floor or table on which the patient is lying. The head must not be allowed to move while the needle is inserted. The needle is inserted at 1 to 2 cm caudal to a point corresponding to the intersection of the dorsal midline, and a line is drawn between the cranial aspects of the wings of the atlas. This point is usually 6 to 9 cm ( In most large animals the needle is seated at approximately 5 to 8.75 cm (2 to Ultrasonography can be used as an aid to needle placement for collection of CSF from both the atlantooccipital cistern and the lumbosacral subarachnoid space.13,14 Contrast material for myelography is injected into the cisterna magna in large animal patients. Withdrawal of CSF before injecting the contrast medium is unnecessary, as is reinjection of CSF after it has been withdrawn. The turnover of CSF is rapid and under strict homeostatic control; therefore withdrawal of CSF through a spinal tap does not have deleterious effects that require its replacement.15–17 The color of the CSF should be noted as it flows from the hub of the spinal needle. Blood can originate from the tapping procedure (iatrogenic hemorrhage) or from a traumatic CNS lesion. Iatrogenic hemorrhage is unevenly mixed in the CSF and disappears as the fluid drips from the needle. Fluid collected immediately after placement of the spinal needle tends to be mildly contaminated with blood even when this is not apparent grossly. Successive aliquots are usually less contaminated, so the later aliquots are most suitable for cellular and protein analysis.18 Blood resulting from CNS hemorrhage is evenly mixed with CSF even after a large amount has been removed. Hemorrhage that has occurred days earlier may have a brownish rather than red discoloration. Prior hemorrhage also results in xanthochromia, a yellow discoloration of the CSF. Xanthochromia can be observed in the CSF for at least 10 days after the introduction of blood. Xanthochromic samples do not contain bilirubin. The normal values for CSF are presented in Table 35-1. Cell counts should be determined in a noncentrifuged specimen as soon as the sample is collected, using a hemacytometer. Automated methods for counting cells are not suitable for CSF because the low numbers of cells in CSF compared with blood result in erroneous counts. Morphologic examination of cells from CSF is most suitably done on cytospin preparations in an appropriately equipped laboratory. Normal CSF from large animals contains fewer than six white blood cells (WBCs) per deciliter. Some reports have documented occasional WBC counts greater than 40 cells/dL in the CSF of normal horses.19 Although suggested to correct for the effects of iatrogenic blood contamination, various “correction factors” are inaccurate and should not be used.18,20 TABLE 35-1 Normal Range of Values in Cerebrospinal Fluid for Large Animals1,4,5 * Cerebrospinal fluid (CSF) sodium is the same as serum or plasma sodium when measured with ion-specific electrodes. When other measurements are used, the CSF sodium is usually higher than plasma sodium. NA, Result not available. The refractive index of normal CSF is less than 1.335. The protein concentration of CSF in normal adult ruminants is less than 50 mg/dL, and in normal horses it is less than 100 mg/dL, although reference values vary with the techniques used for protein measurement. Reference values should be established for each laboratory.21 Studies of the specific and relative quantities of albumin and immunoglobulins, particularly IgG and IgM, have been stimulated by the increasing importance of immunologic testing of CSF for diseases such as equine protozoal encephalomyelitis. Reference values for a number of parameters have been established.22 The most important of these parameters are albumin quotient, a measure of blood-brain barrier (BBB) permeability,23 and IgG index, a measure of intrathecal production of IgG.24 The formulae for these parameters are as follows: Reference values must be established for each species. An elevated albumin quotient indicates BBB leakage or contamination of CSF with blood, with possible introduction of immunoglobulins from serum. An elevated IgG index indicates intrathecal production of IgG and may support a diagnosis of infectious disease of the CNS. The usefulness of these parameters is influenced by a number of other variables, including the immunoreactivity of blood.24 Therefore clinical interpretations must be made with great caution. The composition of protein from the CSF of horses and cattle has been established.3–5,10 The concentrations of glucose and protein in the CSF of newborn foals are almost twice those found in the CSF of adults, but they approximate adult normal values by 2 weeks of age.25 In contrast, calves between 1 and 2 months have lower protein concentration and higher leukocyte numbers in CSF than adult cattle.26 Neural tissue contains the BB isoenzyme of creatine kinase (CK), which increases after damage to the nerve cells.27,28,30 The molecule does not cross the BBB, so CK in the CSF originates from neural tissue. Contamination of CSF with dura or fat, however, falsely elevates the CK concentration. Some have suggested that CK is an accurate marker and a prognostic indicator for CNS disease, but other studies have not supported this conclusion, and measurement of CK, although interesting, has limited clinical utility.27,29,30 The normal CSF concentration of glucose is approximately 80% of that in blood. A decline in the ratio of CSF to serum glucose occurs in animals with bacterial meningitis because of increased use of glucose by inflammatory cells. Measurement of the sodium concentration of the CSF may be helpful for diagnosing salt poisoning in cattle. In animals that do not have salt poisoning, this value is less than 160 mmol/L; in animals with salt poisoning, the concentration is usually greater than 160 mmol/L. Robert J. MacKay The causative agents of eastern, western, and Venezuelan equine encephalomyelitis (EEE, WEE, and VEE) are New World arboviruses belonging to the genus Alphavirus of the family Togaviridae.1 Ross River virus is an Old World alphavirus in Australia that has been shown to cause fever in some experimentally infected horses and is suspected of causing a syndrome of lethargy, muscle soreness, and ataxia in horses in Victoria and South Australia.2 EEE, WEE, and VEE affect all common domestic equid species including horses, mules, and donkeys. Each of the viruses was first isolated during epizootics in the 1930s.3–5 Alphaviruses are unsegmented, single-stranded, positive-sense ribonucleic acid (RNA) viruses; viral genomes are 11 to 12 kB, and infectious virions are spherical enveloped particles 60 to 70 nm in diameter.6 Each of the 3 equine alphaviral encephalitides is among the 13 equine infectious diseases that are notifiable to the World Organization for Animal Health (Office International des Epizooties [OIE]). Data on the occurrence of these diseases in the United States are collected by veterinarians, diagnostic laboratories, and state animal health officials and reported to the National Animal Health Reporting System. At least three subtypes of WEE have been identified. Two of these are found in both North and South America, suggesting that circulation of viruses between the New World continents is common.6,7 There is a distinct North American variety and three South American antigenic varieties of EEE.6 In contrast to the situation with WEE, there is no evidence for movement of EEE viruses between North and South America. The VEE antigenic complex contains 8 viral species, 7 subtypes, and at least 14 varieties,6,8 most of which are maintained continuously in enzootic life cycles in northern South America. Until the 1990s, when an IE variant caused two outbreaks in Mexico, all equine epizootics had been caused by subtypes IAB and IC.9 What was almost certainly the first recorded outbreak of WEE in horses occurred in Kansas, Nebraska, Colorado, and Oklahoma during the late summer and early fall of 1912 when at least 35,000 horses died of the disease.4,10 Epizootics in California and other western states in 1930 to 1932 resulted in the deaths of an estimated 9200 horses and mules (≈50% of those affected).4,11 In 1938 WEE was recognized in every state west of the Mississippi River and affected more than 184,000 horses in that year alone.12 By the 1970s, WEE had been recorded as far east as Tennessee. WEE virus was also implicated in outbreaks of fatal equine disease in the early 20th century in Canada and Argentina, and epizootics have since been reported in Saskatchewan, Alberta, and Manitoba in western Canada; Mexico; Central America; and South America.13 Between 1972 and 1981 there was an annual average of 267 cases of WEE in horses reported by the National Veterinary Services Laboratory in Ames, Iowa12; in 1993 there were 15 cases in 10 states, and since 2005 no case has been reported in the United States. There have been isolated cases of encephalomyelitis in Florida horses associated with the Highlands J virus, an eastern member of the WEE virus complex with epidemiologic characteristics similar to those of EEE virus.14 An outbreak of what was probably EEE was recorded in 1831 in Massachusetts.15 Approximately 100 horses were affected and 75 died. Over the subsequent century, there were at least five other outbreaks consistent with EEE—in Long Island, New York (1845); North Carolina (1902); New Jersey (1905); Florida (1908); and Maryland, New Jersey, and Virginia (1912).16 In 1933 there was an epizootic involving a least a thousand horses in the coastal regions of Delaware, Virginia, and Maryland.3 Epizootics of EEE in North America have since occurred periodically in the coastal areas of the Atlantic, the southeastern United States, and Texas. The largest recorded outbreak was in 1947 in southern Louisiana and Texas when 14,344 cases of EEE were recorded, resulting in 11,722 deaths (mortality rate of 81%).17 Focal outbreaks and sporadic cases have also occurred more or less regularly in Michigan and are occasionally reported from other locations in the eastern half of the United States and Canada. A single case of EEE in a stallion in California was reported in 2002.18 Although the origin of the infection in this horse was not determined with certainty, incompletely activated EEE vaccine was suspected. Despite the availability of effective vaccines since the late 1930s, cases of EEE still occur every year in Florida and other southeastern states. The annual incidence of EEE in horses in the United States over the past 10 years has ranged from 71 (2011) to 732 (2003). Epizootics of what was probably EEE have been reported in Argentina since 1908. Since then, reports of EEE in Central and South America have been widespread, extending from Panama south to Argentina.16 The disease has also caused serious losses in the Caribbean with epizootics documented in the Dominican Republic, Haiti, Jamaica, and Trinidad.15 The disease in equids was first recognized and described in Venezuela and Colombia in 1938.5 At intervals of approximately 10 years, there were large subtype IAB or IC epizootics in northern South America that usually involved tens of thousands of horses. Some of these outbreaks may have been caused by incompletely inactivated vaccine. One epizootic in central Colombia during 1962-1964 was estimated to have killed 100,000 horses.19 An outbreak of VEE caused by subtype IAB began in Peru in the winter of 1969, jumped to Central America in June 1969, and then entered Mexico in 1970. The outbreak spread north to the U.S. border in the spring of 1971, and the first case of VEE in a U.S. horse was confirmed on June 30.20 By the time of the last case on November 7, 1971, more than 1500 horses had died in the southern counties of Texas. There was no further epizootic activity reported until 1992, when a series of epizootics of subtype IC VEE began. Subtype IE was associated for the first time with fatal VEE in small epizootics in Mexico in 1993 and 1996.21 In North America, the WEE and EEE viruses are maintained between epizootics by low-level cycling between New World passerine birds (i.e., songbirds) and ornithophilic mosquitoes in freshwater, forested swamp habitats.1 Snakes may also be an overwintering host for North American EEE virus.22 Infected mosquitoes can be found throughout the year in Florida and other southeastern states. Under favorable environmental conditions, the viruses periodically spread outward from focal reservoirs to infect the general wild bird population, where they are spread and amplified by rapid bird-mosquito-bird transmission. The mosquito vectors involved in maintenance and amplification of WEE and EEE viruses are usually Culex tarsalis and Culiseta melanura, respectively. Many avian species including migratory passerine birds (e.g., starlings and northern cardinals) and wading birds become infected and develop high-order viremia and high serum titers to EEE or WEE but usually do not become ill. Some introduced species, including pigeons, house sparrows, Chukar partridges, and Chinese pheasants, may suffer high morbidity and mortality when infected with EEE virus. Cases of encephalitis usually begin in susceptible horses 2 to 3 weeks after the virus spreads into birds. Human cases may occur several weeks later. Culex tarsalis transfers WEE virus from bird to horse (and human). Although C. melanura may also function in some locations as a bridge vector for EEE, mosquitoes of a variety of other species usually transfer the virus from bird to horse.6,16 Although mosquito-mediated transmission of EEE virus between horses has been confirmed in an experimental setting,23 viremia is low titer, so further infection of feeding mosquitoes with either EEE or WEE virus is unlikely and horses are considered “dead-end” hosts. Both EEE and WEE viruses have been associated with naturally occurring and experimentally induced neurologic disease in calves, and EEE has been associated with fatal infections in New World camelids, deer, ratite birds, cats, dogs, mice, foxes, sheep, and pigs.6,24–26 Epizootics of EEE and WEE usually last 1 to 3 months and occur in summer and early fall when warmth and humidity favor breeding, longevity, and mobility of mosquito populations.27 Outbreaks in Massachusetts and Michigan were most likely during the second consecutive year in which rainfall exceeded the annual mean by more than 20 cm; however, unusually high temperatures did not appear to be a risk factor.27,28 The month of peak incidence of disease onset varies from June in Florida to August or September in northern and western states.4,24,29,30 In Florida and some other southeastern states, isolated cases of EEE can be seen at any time of year.29 Standing surface water for mosquito larval development, bush cover for wild hosts, and the immune status of the various hosts also affect the timing and magnitude of equine epizootics. Many of these physical factors are significantly affected by the cultivation, clearing and irrigation of land, and by drainage of swamps. The proximity and size of plantations of trees were risk factors for EEE for horses in Florida.31 The equine epizootic usually declines with the onset of cool or dry weather unsuitable for mosquito and/or bird activity, as well as the depletion of susceptible equine hosts by death or development of immunity among survivors. The epidemiology of EEE and WEE viruses in South America is poorly understood. Small mammals play a greater role in sylvatic cycles of viral transmission than is the case in North America. Culex (Melanoconion) subspecies and Ochlerotatus albifasciatus appear to be involved in transmission of EEE and WEE, respectively.7,32 Enzootic VEE viruses are maintained by cycling between mosquitoes and small rodents. The Everglades virus, a subtype II found in the Everglades region of Florida, is of this type, cycling between Culex (Melanoconion) subspecies mosquitoes and wild rodents.8 The epizootic varieties IAB and IC have probably arisen on at least four separate occasions by mutation of a single enzootic subtype 1D strain and changes in host range.9 Because of the high-order viremia that develops in infected animals, the defining characteristic of epizootic VEE varieties is the use of equids as amplification hosts. Domestic rabbits, goats, dogs, and sheep also suffer fatal disease during VEE outbreaks.8 There does not appear to be any single “epizootic” vector mosquito species; during outbreaks, virus may be isolated from mosquitoes of many different species.8 Epizootics terminate when susceptible horses are no longer available. The leapfrog spread characteristic of VEE is usually attributed to transport of horses incubating the virus. Movement of infected bats may also contribute to discontinuous spread of virus. By contrast to the situation with the WEE and EEE viruses, birds do not have an important role in amplification and spread of epizootic VEE virus. It is not yet clear whether neurovirulent subtype IE virus is amplified in horses. Evidence from experimental infections suggests that viral titers in blood of infected horses may be insufficient to infect feeding mosquitoes.33 The interepizootic reservoir of epizootic varieties, if any, is not known. Clinical signs following infection with each of the viruses are similar, although signs of EEE are typically more severe and rapidly progressive than those of VEE and WEE.* Infected horses respond in any to all of the following ways after experimental infections36: (1) Inapparent infection with a very-low-grade viremia and fever about 2 days after inoculation. This presentation corresponds to the initial viremia following viral proliferation in regional lymph nodes and probably occurs commonly during outbreaks without progression to subsequent stages. (2) Generalized febrile illness (up to 41.7° C), with anorexia, depression, tachycardia, and diarrhea. This stage is associated with viral proliferation in various body organs after spread from regional lymph nodes. (3) Clinical encephalomyelitis, which is the classic form of the disease. Young horses (especially yearlings) are most susceptible: Approximately 90% of 95 horses with EEE admitted to the University of Florida (UF) were younger than 5 years of age. The onset of neurologic signs, associated with the second febrile crisis, typically occurs about 5 days after infection (range 2 days to 2 weeks), and most deaths occur 2 to 3 days later.35,36 Almost all horses with EEE are febrile at the onset of neurologic signs (mean of 40.3° F in horses admitted to UF), although the temperature declines thereafter. The onset of severe neurologic signs is peracute to acute, and progression is rapid. Horses may be found recumbent and comatose or dead. If observed, initial CNS signs are quite variable and referable to diffuse or multifocal cerebral disease; evidence of brainstem and spinal cord involvement quickly becomes obvious as the illness progresses (Fig. 35-3). Obtundation is the most common initial clinical sign, although owners also report suspected colic as part of the initial presentation. Affected horses often stand apart from pasturemates, have no interest in food, or fail to respond to an owner’s call. Further signs of dementia may follow including head pressing, teeth-grinding, hyperesthesia, irritability, aggression (rarely), leaning against a wall or fence, or compulsive walking, often in a circle (especially around the inside of a stall or small paddock). Several observers have noted a “peculiar looseness of the lips” as a characteristic early sign.4,37 Blindness and lack of a menace response may be noted at this stage. Signs of cranial nerve disease including abnormal pupillary light reflexes, head tilt, nystagmus, tight circling, facial and tongue paralysis, and inability to swallow often occur as the disease advances. Abnormal signs usually present asymmetrically, at least initially. Ataxia and paresis of the trunk and limbs result in a progressively more unsteady gait. Tremors are seen in the antigravity muscles of the limbs. Once recumbent, affected horses seldom regain their footing and are often noted to make galloping or swimming movements in lateral recumbency.4 Seizures occur in about a third of horses with EEE and may happen at any stage. Mucopurulent nasal and ocular discharges and abrasions/lacerations of the face and limbs are the most common non-neurologic findings; the latter occurs secondary to trauma from falling, seizures, or running into objects. Mortality rates are 75% to 95% for EEE, 19% to 50% for WEE, and 19% to 83% for VEE.4,6,17,32,38 Vaccination within the preceding year was associated with lower risk of death in horses with EEE.27,29 Death is usually preceded by a period of recumbency during which the horse may be semicomatose and convulsing. Surviving horses gradually recover over a period of weeks. Many horses with WEE apparently recover completely, but it is estimated that two thirds of horses that survive EEE have residual signs of CNS damage such as central blindness, dullness, and diminished learning capacity or signs of cranial nerve dysfunction such as facial paralysis and head tilt.16 Such horses are often referred to as “dummies.” Abnormalities seen in hemograms and plasma chemistry panels of horses with alphaviral encephalomyelitis are minor and nonspecific. Horses with EEE may have abnormal WBC counts and high fibrinogen concentration. In 73 horses with EEE admitted to the University of Florida, the following hematologic abnormalities were noted: leukocytosis (56%), neutrophilia (60%), hyperfibrinogenemia (57%), monocytosis (36%), lymphopenia (13%), leukopenia (7%), neutropenia (4%), and lymphocytosis (3%). Slight leukopenia was noted during experimental infections of horses with WEE and VEE viruses. Mild elevations in plasma enzyme activities of affected horses reflect organ damage secondary to anorexia, fever, stress, recumbency, and seizures. Hyponatremia is found in some horses with EEE; in humans with EEE, hyponatremia predicts mortality but its significance is unknown in horses. The results of CSF analysis for horses with EEE are quite distinctive. More than 95% of horses with EEE admitted to the University of Florida had abnormal CSF cytology. Among UF patients, 88% of samples had high protein concentrations (mean of all samples: 139 mg/dL, range 38 to 526 mg/dL). Nucleated cell counts were high in 87% of samples (mean 248 µL, range 0 to 3100/µL; normal range 0 to 6/µL); pleocytosis is characterized by high neutrophil concentrations (mean 46%, range 0 to 97%). Neutrophils are typically nondegenerate. Protein concentration and nucleated cell counts are typically higher in samples obtained from the atlantooccipital (AO) site than they are in samples from lumbosacral taps. In the WEE outbreaks of 1930 to 1932, CSF nucleated cell counts were typically 12 to 30/µL and most cells were lymphocytes.11 A presumptive diagnosis is usually made clinically, especially in areas where these diseases are prevalent during the mosquito season. The finding in a CSF sample of moderate neutrophilic or lymphocytic pleocytosis further supports a diagnosis of alphaviral encephalomyelitis. Hemagglutination-inhibition (HI), complement fixation, fluorescent antibody, and virus neutralization tests have traditionally been used for serologic diagnosis.16 The immunoglobulin M antibody capture (MAC) enzyme-linked immunosorbent assay (ELISA) is the method of choice for use in horses with viral encephalomyelitis because at titers of greater than 1 : 400 it can distinguish between vaccinal (IgG only) and viral infection–induced (IgM and IgG) titers.33,39 These assays are available at many state diagnostic laboratories and also at the National Veterinary Services Laboratories (NVSL) in Ames, Iowa. In surviving horses, demonstration of a fourfold or greater rise in HI or plaque-reduction neutralizing titer between acute and convalescent samples provides additional diagnostic support. It should be noted that positive results in these tests could simply reflect subclinical infection with either virus. Virus can be isolated from fresh or frozen brain in tissue culture or mice and can be detected in histologic sections by immunohistochemistry or polymerase chain reaction (PCR) techniques.40 Differential diagnoses include hepatoencephalopathy; gastrointestinal hyperammonemia; heavy metal or other toxicosis; rabies; other viral encephalomyelitis (West Nile, Borna disease, equine herpesvirus myeloencephalopathy [EHM], Aujeszky disease, louping ill); equine protozoal myeloencephalitis (EPM); trypanosomal (South America), verminous, or bacterial meningoencephalomyelitis; space-occupying lesion within the calvarium (e.g., cholesterol granuloma, neoplasia, brain abscess); brain infarct; brain trauma; and leukoencephalomalacia. Treatment is largely supportive and, in the case of EEE, usually ineffective. Any adult horse with EEE that is unable to stand should be euthanized. Such horses should not be subjected to slinging. Hyperthermia is known to exacerbate brain injury and must be treated vigorously with cold water or alcohol baths until rectal temperature is less than 102° F. Convulsions can be controlled with diazepam, xylazine, or barbiturates (pentobarbital or phenobarbital). In addition to excellent general nursing care, all horses should be treated for brain edema and inflammation, although only horses whose signs are mainly cerebral are likely to respond. Drugs used for this purpose include dimethyl sulfoxide (DMSO), 1 g/kg as a 10% solution intravenously (IV) or intragastric, flunixin meglumine (or equivalent nonsteroidal antiinflammatory drug [NSAID]) intramuscularly (IM), IV, or orally (PO) at 1.1 mg/kg q12h for 3 days, dexamethasone (or equivalent corticosteroid) IM or IV at 0.05 to 0.1 mg/kg daily for several days for 3 days then tapered, and mannitol (IV at 0.25 to 1 g/kg as a 20% solution) or hypertonic saline (e.g., 7.2% saline IV at 2 mL/kg 4 to 6 times during the first day). A lengthy course of dexamethasone is believed anecdotally to be an important determinant of neurologic recovery in surviving cases. Treatment should be titrated to minimize neurologic signs but generally must be tapered over several weeks. Complete recovery, if it occurs, may take several months. Treatment with serum from horses hyperimmunized against WEE virus was thought to reduce mortality modestly during the 1930 to 1932 outbreak; however, these claims were not tested scientifically.4 Furthermore, administration of large quantities of hyperimmune serum to experimentally infected horses by investigators at Lederle Laboratories was reported to have no beneficial effect.41 Adjuvant antiviral therapy with interferon (IFN) α or β has salutary effects in some experimental models of alphaviral encephalitis42 and could be tried in valuable horses. A reasonable protocol is described in the West Nile encephalitis section. The nucleoside analogue ribavirin boosts the antiviral activity of IFN in vitro against encephalitic alphaviruses43 and holds some potential as future therapy for EEE in humans and perhaps horses. All three viruses infect humans causing subclinical, febrile, or neurologic disease.6 Since 1964, there have been 640 cases of human WEE reported in the United States, with an average of 27 cases/year in the period up to 1987. Since 1988, there have only been five cases of WEE reported. By contrast, the 268 cases of EEE in humans reported in the United States since 1964 have occurred at a fairly steady rate of 6 cases/year. Epidemics of VEE are closely associated with equine epizootics; thus during the 1962 to 1964 epizootic in central Colombia, which killed about 100,000 horses, 200,000 humans were infected, of whom 0.5% died. Although nonpathogenic for horses, the enzootic Everglades subtype II strain has caused human encephalitis in Florida. No cases of human disease were recorded during the VEE subtype IE equine epizootics in Mexico in 1993 and 1996, and South American strains of EEE are not known to cause encephalitis in humans. Short-term mortality rates of EEE, WEE, and VEE in humans are 74%, 4%, and less than 1%, respectively. Most survivors of EEE suffer long-term neurologic impairment. There is congestion of most organs and a slate-gray discoloration, often with petechial hemorrhage, of the brain and spinal cord, especially obvious on section of formalin-fixed tissue.40,44,45 Often there is brain swelling and evidence of occipital subtentorial herniation, with brainstem compression. Histologically, there is evidence of acute to subacute and multifocal to diffuse meningoencephalomyelitis. There is predominant involvement of gray matter, with diffuse neuronal degeneration, gliosis, perivascular and neuroparenchymal infiltrates, and meningitis. Neutrophils are prominent in acute EEE, whereas lymphocytes predominate in older lesions. On occasion, eosinophils may predominate in EEE. Lymphocytes are usually the primary inflammatory cell in WEE lesions.44 Previous therapy with potent antiinflammatory agents, particularly corticosteroids, may suppress the inflammatory-cell infiltrates, which then appear mild. Bronchopneumonia is found in about 20% of fatal cases of EEE, probably secondary to pharyngeal paresis and aspiration of particulate material. Up to 10% of horses with EEE have histologic evidence of myocardial necrosis, likely a sign of brain-heart syndrome.45 Because high infection rates in humans have been documented following exposure to aerosols from infected laboratory animals or from laboratory accidents (or, potentially, equine carcasses), necropsies of horses with suspected VEE should only be performed by personnel who possess demonstrable immunity in the form of neutralizing antibody.46 All laboratory manipulations must be carried out within certified biological safety cabinets following containment level 3 procedures. Active immunization through use of formalinized vaccines began in 1934 and is still the only commercial method of immunization of horses against EEE and WEE. These inactivated vaccines are also used routinely to protect ratites from EEE and WEE and South American camelids from EEE.47 The attenuated, tissue-culture-origin vaccine, TC-83, derived from a Trinidad VEE virus isolate, which was originally developed to protect military personnel, provides durable immunity against VEE and was effective in minimizing the incursion of VEE into the United States in 197120 and in limiting the 1993 and 1996 outbreaks in Mexico. Although TC-83 is no longer licensed for use in horses in the United States, a formalin-inactivated vaccine containing the TC-83 vaccine strain is available and is often included with EEE and WEE in trivalent products.48 For any of the formalin-inactivated alphavirus vaccines, adult horses should be given an initial two inoculations 4 to 6 weeks apart at least a month before the anticipated risk period or before the onset of vector mosquito activity and then revaccinated at intervals reflecting the likely risk. Vaccination should begin in January or February in Florida to May or June in parts of Canada and the northern United States. Clinical evidence suggests less than 6 months of protection following vaccination against EEE or WEE, so revaccination at least once during the mosquito season is necessary in areas with warm climates and long mosquito seasons. Vaccinated mares should receive a booster dose 4 to 6 weeks before foaling to provide colostral antibody for newborn foals. Foals should be given an initial inoculation at 4 to 6 months (vaccinated mare) or 3 to 4 months (unvaccinated mare), then a second inoculation 4 to 6 weeks later and a booster at 10 to 12 months of age. AAEP guidelines for 2012 recommend that foals of vaccinated dams in high-risk areas should be given a primary series of four doses beginning at 2 to 3 months of age. Two additional inoculations are then given at approximately 4-week intervals and a booster at 10 to 12 months of age, before the onset of mosquito season. It will be important to validate this method of vaccination because of concerns that early frequent vaccinations may suppress immune responses to EEE.49 Until this issue has been resolved, the author will continue to recommend that the initial inoculation be given to foals in high-risk areas no earlier than 4 months of age (as for foals in moderate-risk areas). Routine surveillance of the virus pool by serum antibody testing of sentinel chickens and virus detection in trapped mosquitoes by health authorities often provides early warning of an impending outbreak and allows time for vaccination of susceptible horses. General mosquito control (removal of standing water, spraying by local authorities) will reduce risk of EEE and VEE in horses.27 Because of the role of horses in amplification and transmission of VEE virus, quarantine of infected and exposed horses with area-wide and international restrictions on horse movement is an integral part of the management of VEE epizootics.50 Robert J. MacKay Since the encroachment of West Nile virus (WNV) in 1999 into North America, the potential for emerging foreign disease with widespread impact has been realized. Given the opportunity, many other viral infections could become established in new countries as a result of the globalization of the horse industries and effects of global warming. The Semliki Forest complex of the Togaviridae are widespread throughout their geographic range, either in the Orient (Getah) or South Pacific (Ross River).1,2 Although closely related, there is little geographic overlap among these viruses. As with the North American alphaviruses, these viruses are transmitted by mosquitoes (Aedes, Anopheles, and Culex spp.).3–6 The reservoir for Getah virus (GV) is not known, although swine appear important in its transmission to mosquitoes.6,7 Other feral species are not firmly established. For Ross River virus (RRV), marsupials are important reservoirs.8 With GV, horses might develop sufficiently high viremia to transmit virus, but this is unlikely with RRV. Many animals have been found to be seropositive to GV, including ruminants, and seroprevalence in horses varies from less than 10% to more than 90%.6,9–11 RRV is endemic in horses and widespread, with seroprevalence ranging from 50% to 80% in Australian horses.1,12–14 With the recent increase in Kunjin/West Nile encephalitis in Australia, RRV has become an important differential diagnosis for horses with neurologic disease and is usually included in serologic and PCR testing of diagnostic samples. Getah virus is likely more pathogenic in horses than RRV.9,11,15 Clinical signs include pyrexia, edema of the limbs, and stiff gait. Horses may also develop urticaria and submandibular lymphadenopathy. The course of GV disease is 7 to 10 days. With RRV, horses also develop fever, lameness, swollen joints, inappetence, generalized stiffness, and even colic.1,12,14 High rates of mortality are not a feature of either virus. Serologic detection of IgM is the mainstay of diagnosis of these viruses, although a vaccine against GV complicates diagnosis.16,17 RRV can be detected by PCR and isolated from the blood of horses during the disease course.18 Pathologic changes associated with these viruses are seldom described because of the low mortality of associated diseases. Horses primarily develop perivascular inflammation in the brain. Treatment is supportive; no antiviral is specific for alphaviruses. A vaccine for GV is available in Japan. An inactivated vaccine against RRV for humans is being tested in Australia, but no equine vaccine is available.19 Classical Borna disease (cBD) is a viral encephalitis principally of horses and sheep that was first recognized in southwestern Germany in the 18th century but is associated with the village of Borna in the Kingdom of Saxony, where the disease emerged in the late 19th century. It is estimated that approximately 16,000 horses died annually in Saxony from 1896 to 1940.20 These numbers declined rapidly in the 1960s in parallel with declining horse numbers, and only 1216 horses died of cBD in Saxony from 1958 to 1991.20 Outbreaks of cBD have been restricted to central Europe in the upper Rhine valley around Switzerland, Austria, and Liechtenstein. Borna disease virus (BDV)-associated encephalitis has also been reported in cattle, an alpaca, deer, zoo herbivores, cats, rabbits, and dogs.21 During recent decades, evidence of BDV infection including antibodies, antigen, RNA, or the virus itself has been reported in many countries and in numerous mammalian species including humans. There is serologic evidence of BDV infection in horses in the United States.22,23 Some of these reports are questionable because of problems with contamination of samples by laboratory BDV strains and poorly validated immunoassays; however, there are enough well documented infections to confirm widespread distribution of BDV or BDV-like virus.21 The possibility of involvement of BDV in several human neuropsychiatric diseases has raised interest in BDV but remains controversial. The etiologic agent of Borna disease is a nonsegmented, negative-sense, single-stranded RNA virus of the order Mononegavirales, family Bornaviridae. Although regional genetic variation occurs, the BDV genome is considered remarkably stable for an RNA virus. Related viruses, termed avian Borna viruses (ABVs), have been discovered in birds, and endogenized BDV sequences are common in the genomes of mammals. The significance of this last intriguing finding is unknown. Most outbreaks in horses and sheep occur during the spring and early summer. Although horse-to-horse transmission is theoretically possible, it is unimportant in the evolution of an outbreak.21 The virus is likely maintained in small mammals with geographic ranges approximating the endemic Borna disease area. Bank voles, bicolored white-toothed shrews, and bats have been suggested as possible reservoir hosts.24 BDV is shed through nasal and lacrimal secretions and in the urine of infected animals.21,25 BDV is resistant to drying and other adverse environmental conditions. It is assumed that BDV enters the horse though intranasal infection and migrates to the brain transaxonally.26 Direct replication occurs in neurons and glial cells, with centrifugal spread to peripheral nerves and retina. BDV infection of the retina may contribute to the blindness that characterizes acute Borna disease in horses.27 Subclinical BDV infection is common with seroprevalence of horses in endemic areas of 10% and 20%, and as high as 50% among stablemates of horses with Borna disease. The neurologic signs of Borna disease are associated with the florid cell-mediated initial immune response to the virus. In subclinically infected horses and horses that recover from Borna disease, the virus becomes quiescent and largely undetected by the immune system. The clinical signs of Borna disease in large animals are similar to those of the other equine encephalitides. The incubation period is thought to be about 2 months in naturally occurring cases. There is a predominance of forebrain signs initially, followed by signs of brainstem and spinal cord involvement. In moderate to severe cases, horse die 1 to 4 weeks after onset of disease. In mild cases, recovery can occur, but persistent abnormalities and exacerbations are common. There is a prodrome characterized by fever, inappetence, colic, and vague behavioral changes. The onset of neurologic signs is marked by changes in mentation with alternating hyperresponsiveness and stupor; compulsive walking, often in circles; teeth-grinding; and aggression. Other signs of brain disease include trismus, pupil constriction, nystagmus, head tilt, and blindness. With progression, there is ataxia and weakness of the limbs and trunk and areas of cutaneous desensitization or hyperesthesia. Collapse into recumbency and seizures are the final stages. The mortality rate is about 80% in horses and 50% in sheep. For antemortem diagnosis, antibodies to the agent may be found in the serum and CSF of most infected animals by ELISA, indirect fluorescent antibody (IFA), and Western blot.24 CSF analysis often reveals mononuclear pleocytosis and high protein concentration. Reverse transcriptase nested PCR techniques have demonstrated virus in the CSF and peripheral mononuclear cells.28 The criteria for diagnosis include a horse with neurologic abnormalities testing antibody positive for BDV in serum or CSF or a horse with neurologic signs and appropriate histopathologic changes. Pathologic abnormalities resemble those of a viral encephalomyelitis, and confirmatory diagnosis is by immunohistochemistry, virus isolation, and detection of viral nucleic acids.21,24,29–33 The characteristic microscopic lesion of Borna disease is the Joest-Degen inclusion body in the neuronal nucleus, but this is not always observed. Virus is detectable easily with monoclonal or polyclonal antibodies. Histopathologic changes are those of a typical polioencephalomyelitis, with a particular loss of neurons in the hippocampus. There is involvement of the gray matter of the olfactory bulb, basal cortex, caudate nucleus, thalamus, hippocampus, and medulla oblongata. No antiviral agents are available for BDV, and use of the amantadine sulfate (developed for treatment of influenza infection) is controversial but commonly used.34,35 Likewise, vaccination as a protective strategy is controversial. It is widely assumed that a modified live vaccine would offer more protection; however, the live vaccine available until the early 1990s in East Germany was removed from the market because of questionable efficacy. Hendra virus (HeV) infection of horses was first recognized in 1994 during simultaneous outbreaks of severe respiratory disease in Queensland, Australia: one in the Hendra suburb of Brisbane and the other near the town of Mackay. Mortality was high among the 22 affected horses and each outbreak was associated with transmission to humans, 2 out of 3 of whom died.36–38 Since 1994, there have been at least 33 outbreaks of HeV infection in horses, all in Queensland or northern New South Wales. Cases have occurred every year since 2006, and there were 11 outbreaks involving 23 horses in 2011. Five outbreaks also involved seven human cases, of which four were fatal. Signs of neurologic disease were seen in some horses during the initial outbreaks, and neurologic signs dominated the clinical presentation in five horses in an unusual outbreak at an equine referral practice in the Redlands suburb of Brisbane in 2008. The signs were those of fulminant febrile encephalitis with severe obtundation, dementia, vestibular signs, and progressive ataxia. Nipah virus (NiV) emerged as the cause of porcine respiratory and encephalitis syndrome in Malaya in 1998.38–40 Signs were predominantly respiratory, and most affected pigs recovered. NiV also caused severe febrile encephalitic illness among workers in contact with infected pigs; 105/265 affected people died. NiV has also been recovered from the brain of a horse with encephalitis. HeV replicates initially in the respiratory epithelium before spreading hematogenously to multiple organs, including the CNS, to cause widespread vasculitis, endothelia syncytia, vascular thrombosis and hemorrhage, and cellular necrosis. HeV and NiV are closely related viruses that occupy a new genus, Henipavirus, of the family Paramyxoviridae. They are enveloped, single-stranded, negative-sense RNA viruses. The henipaviruses are highly pathogenic for a wide range of mammals including horses, pigs, and humans. The natural reservoirs throughout the geographic range in Northern Australia, Malaysia, and Bangladesh are fruit bats from the genus Pteropus (also known as flying foxes).41 Transmission from bats in each spillover event is likely via contact of the horse with urine or reproductive fluids from infected bats. Horses incubating HeV are infectious to other horses for at least 48 hours before the onset of clinical signs, and at least two outbreaks in stables were caused by introduction of preclinical horses incubating the virus. Diagnosis is made both by detection of serum neutralizing antibodies by immunoassay and detection of RNA by quantitative reverse transcriptase PCR in blood, other body fluids, or from tissues taken at necropsy.40,42,43 Virus is secreted in all body fluids and feces of infected horses, and appropriate precautions must be taken to prevent transfer to other horses and humans. Guidelines for biosecurity have been developed by the Australian Veterinary Association (available at http://www.ava.com.au) and by Biosecurity Queensland (http://www.biosecurity.qld.gov.au). Infected horses are euthanized as a matter of public health. Ribavirin is used with partial success to treat NiV and HeV infections of humans, and a monoclonal antibody (mAb m102.4) has been shown to be protective in experimental animals if given before clinical signs and has been used in several exposed humans.44 An HeV subunit vaccine (HeV-sG) has been tested successfully in multiple species including horses against both HeV and NiV challenge and was launched commercially in Australia in 2013 for vaccination of horses in endemic areas (EquiVac HeV, Pfizer Animal Health).44 The Bunyaviridae are mostly arboviruses and are found worldwide. These viruses are usually maintained in mosquito-vertebrate host-mosquito cycles with midges acting as the arthropod hosts for a few viruses.45 Twelve serotypes, known collectively as the California serogroup, have been isolated in North America, South America, Africa, Europe, and Asia. In rare cases, these viruses can cause acute encephalitis in horses. Snowshoe hare virus and Main Drain virus have been associated with encephalitis in horses,46,47 and serologic evidence of subclinical infections of horses with Jamestown Canyon virus and Cache Valley virus was found in horses in southern California and Michigan, respectively.48,49 Clinical signs in a horse from which Main Drain virus was isolated included ataxia, weakness, stiff neck, head pressing, dysphagia, tachycardia, and fever.47 Equine encephalosis is an arthropod-borne viral infection, affecting all species of equids, usually characterized by mild or subclinical infection but occasionally causing severe clinical disease.50 The causal agent, equine encephalosis virus (EEV), is transmitted through the bites of certain Culicoides spp. biting midges. EEV is an orbivirus, as are African Horse Sickness virus (AHSV) and bluetongue virus (BTV). All equids (horses, donkeys, zebras) are susceptible to EEV, although donkeys and zebras are thought to be resistant to the clinical disease. The infection is endemic in southern Africa where seven EEV serotypes have been identified.51 An outbreak of febrile disease due to EEV in Israel in 2008-2009 involved 60 horses, and serologic evidence of the infection has since been found in several other areas outside southern Africa.52,53 The virus was originally named for isolation from a horse with typical signs of encephalitis including mild to severe ataxia, reluctance to walk and stiffness, dementia, and seizures.50 In its current forms, infections are either subclinical or associated with an influenza-like syndrome of fever, edema, tachycardia, tachypnea, and congested mucosae. An AHSV-like syndrome of respiratory distress and cardiac failure has been described, and pregnant horses may abort. A competitive ELISA for detection of group-specific antibody to EEV has been described for use in diagnosis.54 There is no recognized treatment or method of prevention for EE. Robert J. MacKay The causative agent of West Nile encephalitis (WNE) is a small, positive-sense, single-stranded, enveloped RNA virus.1 West Nile virus (WNV) is a member of the Japanese B encephalitis (JE) antigenic complex of neuroinvasive arboviruses within the genus Flavivirus, family Togaviridae.1 In addition to WNE virus, Kunjin, JE, and Murray Valley encephalitis (MVE) viruses are members of the JE antigenic complex that have caused encephalomyelitis in horses outside the United States. There are two main genetic lineages of West Nile encephalitis virus: Outbreaks of neuroinvasive disease in horses and humans have traditionally been associated with lineage-1 isolates that circulate among Mediterranean basin, West African, and northern European countries. A virus of this lineage is responsible for the North American epizootic. Lineage-2 isolates are widespread in sub-Saharan Africa and commonly have infected horses without apparent clinical effect. In the past few years there have been several cases and even outbreaks of neurovirulent equine and human disease caused by Lineage-2 WNV in Africa and Europe.2,3 The Kunjin virus is a lineage-1 WNE virus that traditionally has caused mostly subclinical or febrile non-neurologic disease in horses and humans in northwestern Australia and Papua New Guinea. A neurovirulent strain of Kunjin virus emerged in 2011 in New South Wales in Australia in the late summer and fall and caused encephalitis in at least 1000 horses in southeastern states with a mortality rate of 10% to 15%.4–6 This unprecedented epizootic of Kunjin encephalitis (and also of encephalitis due to the arboviruses Ross River virus and Murray Valley encephalitis virus) followed unusually high spring rainfalls in southeastern Australia.6 Compared with prototypic Kunjin virus (WNVKUN), the neurovirulent strain (WNVNSW2011) has acquired at least two of the known virulence markers of the North American strain. There was also a sharp increase in Murray Valley encephalitis cases in horses as part of the 2011 arboviral encephalitis epizootic.6 Since the last MVE epizootic in 1974, cases had been reported only sporadically.7 Japanese B encephalitis virus is endemic in east Asia and southeast Asia and has made incursions into the northern tip of Australia.8 It is maintained in cycles among porcine and avian reservoirs using mosquito vector hosts. Humans and horses are dead-end hosts. Cases of encephalitis in horses are seen sporadically throughout the virus’s range, and effective vaccines are available.1 Although there is minimal overlap between the ranges of WNV and JEV, diagnostic serology may yield confusing results in areas where both diseases (and vaccinations) do occur. Louping ill virus and Powassan virus are tick-borne flaviviruses that have the potential to cause encephalitis in horses.4 WNV was originally isolated from the blood of a febrile adult woman in the West Nile region of Uganda in 1937. Cases of encephalitis in humans were first recorded in Israel in the 1950s and in horses between 1960 and 1965 in Egypt and France.9,10 Since 1996 there have been small localized outbreaks of equine WNE in numerous countries in the Mediterranean basin and northern Europe with repeated seasonal epizootics in the Camargue marshlands of France and in the Tuscany region of Italy.11,12 Although the virus usually did not persist in these locations after equine or human disease waned, there is recent evidence for overwintering of WNV in the Tuscany region of Italy.12 In August 1999, the first case of WNE in humans was reported in Queens, New York.13 Characterization of the virus from this outbreak showed it was highly homologous with Israel 1998 (Is98), a strain of WNV that had originally been isolated from a goose.14 Eventually, 62 cases occurred in humans in1999 and 7 died. From August to October 1999, 25 horses on Long Island were diagnosed with WNE.15 Nine of these horses died. Deaths occurred in numerous native bird species, particularly in corvids (crows, ravens, jays) and in exotic species housed at the Bronx zoo. In 2001 the disease became a national epizootic with cases seen for the first time in Florida, Kentucky, Louisiana, Georgia, Alabama, Illinois, Indiana, Mississippi, New Hampshire, North Carolina, Tennessee, and Virginia.16 There were 738 cases reported nationally, of which 33.2% died or were euthanized. Despite the availability of an effective vaccine from August 2001 onward, there was a dramatic westward spread of WNE in 2002, with 15,257 horses affected in 40 states.16 The national incidence of the disease in horses declined in 2003 to 5181 cases in 44 states. By 2006 equine cases had been reported from all 48 contiguous states. Total annual cases dropped below 1000 in 2007, and only 79 were reported in 2011. In 2012 there was an upswing in WNE in all parts of the country with 618 total cases, of which 116 were in Texas.16 The human epidemic dramatically expanded in 2012 with a provisional total of 2734 cases of neuroinvasive disease, second only to the 2866 cases recorded in 2003. This alarming upswing in cases may be explained, at least partially, by the unusually mild winter, early spring, severe drought, and high mean temperatures of 2012. WNV cycles between mosquito vectors and avian reservoir hosts. In temperate climates, the virus overwinters by vertical (transovarial) transmission in vector mosquitoes and possibly by latent infection of avian hosts.1 Culex-species mosquitoes are the most important vectors for the North American epizootic in birds. Different species transmit the virus in different parts of the country. Major vector species include Culex pipiens in the northeast and central United States, Culex tarsalis from the far west to the panhandle of Texas, and Culex quinquefasciatus in the southeastern United States from Texas to Florida. Although birds of more than 300 species have been reported WNV positive, passeriform (true perching bird) and charadriiform (shorebird) species are considered the most competent reservoir hosts for WNV in the United States. Corvid species such as crows and blue jays are efficient hosts but have suffered high mortality during the North American epizootic. The house sparrow (an Old World passerine species) also develops high-titer viremia after WNV infection but is relatively resistant to the adverse effects of the virus. A variety of domestic mammals including sheep, alpacas, and dogs have suffered fatal WNE during the North American epizootic. Horses, humans, and other mammals are “dead-end” hosts and do not amplify the virus in quantities sufficient to infect mosquitoes. In contrast, alligators experience high-titer viremia and may be important reservoirs for infection in the southeastern states. “Bridge vector” species of mosquito transmit the virus from birds to mammals, including horses. The mosquito species important for bird-horse transmission are not known with certainty but may include members of C. pipiens complex, C. tarsalis, and Culex salinarus. The occurrence of WNE reflects vector mosquito activity, which is seasonal in temperate zones and year-round in tropical and subtropical regions. Significant viral activity begins in July, with peak incidence in September and October. The epizootic usually declines rapidly after the first frost. Experimental infections of horses result in viremia 3 to 5 days after viral inoculation into the CSF and clinical signs beginning at 7 to 10 days.1 Experience during clinical outbreaks indicates a similar 1- to 2-week incubation period before signs of WNE are seen. Clinical signs have been extremely varied; however, surveys of cases published across the United States and from Europe have revealed some consistent combinations of signs.9,12,15,17–20 Roughly a quarter of affected horses have had mild to moderate fever at the time of initial examination, although it is likely that most were febrile before examination. Consensus neurologic signs seen have included limb ataxia (71% of 726 cases from the northeastern United States, Florida, Indiana, Nebraska, and Colorado9,12,15,17–20), limb weakness (58%), muzzle twitching (45%), obtundation (43%), recumbency (31%), lip droop (22%), hyperresponsiveness/hyperesthesia (18%), teeth-grinding (12%), dog-sitting (10%), thoracic limb collapse (7%), compulsive walking (7%), muscle atrophy (6%), seizure (4%), blindness (4%), circling (4%), and head pressing (3%). Clinical signs reflect the reported predominance of lesions in the gray matter of the hindbrain and spinal cord of horses with WNE with lesser involvement of the rostral brainstem and forebrain.21 The observation of coarse twitching of the muzzle (and to a lesser extent the eyelids) is a particularly distinctive finding in acute cases of WNE. Less frequently, there are fine fasciculations of the cutaneous muscles of the torso and neck. Horses acutely affected may be frantic, fearful, and hyperresponsive to stimuli. This stage is transient (usually <1 day in duration) and typically is the only behavioral change seen. Mild obtundation is common at initial presentation and progresses to stupor with narcoleptic episodes in horses with severe midbrain or hindbrain involvement. In addition, such cases often have facial and/or tongue paralysis or vestibular signs of head tilt and body lean. Ataxia and weakness of the trunk and limbs occur with or without signs of brain disease and may present asymmetrically. Such presentations are clinically indistinguishable from those of horses with equine protozoal myeloencephalitis. Because of the predilection of WNV for involvement of gray matter in thoracic spinal cord segments, horses may present with the unusual combination of acute-onset bilateral thoracic limb weakness/collapse and normal pelvic limb strength (Fig. 35-4). Severely affected horses are often unable to roll from lateral to sternal recumbency and tend to lie quietly without much voluntary activity. Comparable presentations in human patients with paralytic forms of WNE are described as “polio-like.”22 With time, muscles of the thoracic girdle can become severely atrophied and such horses are rendered permanently unable to move from lateral to sternal recumbency even though they may be able to walk once assisted to stand. The convalescent stage of WNE is described later in the Treatment section.
Diseases of the Nervous System

Cerebrospinal Fluid
Collection of Cerebrospinal Fluid
Lumbosacral Spinal Tap
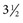 -inch needle can be used in small ruminants, foals, and small cattle). A “snapping” sensation is sometimes felt as the needle passes through the interarcuate ligament. The patient may reflexly contract the tail, anus, and gluteal muscles. Some patients, particularly horses, may respond with violent motor activity. Therefore a lumbosacral spinal tap performed on a conscious horse should be done only when the animal is restrained in stocks. The average depth of insertion is 17.64 cm (7 inches) in horses, 8.26 cm (
-inch needle can be used in small ruminants, foals, and small cattle). A “snapping” sensation is sometimes felt as the needle passes through the interarcuate ligament. The patient may reflexly contract the tail, anus, and gluteal muscles. Some patients, particularly horses, may respond with violent motor activity. Therefore a lumbosacral spinal tap performed on a conscious horse should be done only when the animal is restrained in stocks. The average depth of insertion is 17.64 cm (7 inches) in horses, 8.26 cm (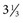 inches) in ponies,10 and about 7.5-10 cm (3-4 inches) in adult cattle.
inches) in ponies,10 and about 7.5-10 cm (3-4 inches) in adult cattle.
Cisterna Magna Tap
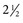 to
to 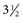 inches) from the poll (Fig. 35-2). A
inches) from the poll (Fig. 35-2). A 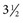 -inch, 18-gauge spinal needle is inserted perpendicular to the skin and aimed toward the nose. The needle is advanced slowly with the stylet seated. After the needle has been advanced a few millimeters, the stylet is removed and the hub of the needle is examined for CSF flow. The stylet is replaced if it is dry and CSF is not spontaneously dripping from the hub, and the needle is advanced another few millimeters and checked again for CSF flow. Entry of the tip of the needle into the cisterna magna may be accompanied by the sensation of “popping” through a tissue plane or by a sudden decrease in resistance to the advancement of the needle. In other cases, however, no such sensation is perceived—thus the precaution of checking for CSF flow every time the needle is advanced a few millimeters.
-inch, 18-gauge spinal needle is inserted perpendicular to the skin and aimed toward the nose. The needle is advanced slowly with the stylet seated. After the needle has been advanced a few millimeters, the stylet is removed and the hub of the needle is examined for CSF flow. The stylet is replaced if it is dry and CSF is not spontaneously dripping from the hub, and the needle is advanced another few millimeters and checked again for CSF flow. Entry of the tip of the needle into the cisterna magna may be accompanied by the sensation of “popping” through a tissue plane or by a sudden decrease in resistance to the advancement of the needle. In other cases, however, no such sensation is perceived—thus the precaution of checking for CSF flow every time the needle is advanced a few millimeters.
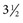 inches). While advancing the needle, the heel of the hand should be held firmly against the animal’s neck to minimize the possibility of spinal cord injury. The mean depth of insertion is 6.16 cm (
inches). While advancing the needle, the heel of the hand should be held firmly against the animal’s neck to minimize the possibility of spinal cord injury. The mean depth of insertion is 6.16 cm (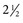 inches) in horses10 and 5.08 cm (
inches) in horses10 and 5.08 cm (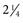 inches) in cattle. In a cisterna magna tap, the needle entry site is close to the cervical spinal cord and the brainstem. To minimize the danger of central nervous system (CNS) damage during a cisterna magna tap, the animal should be adequately anesthetized and ventilated, because an increased partial pressure of carbon dioxide (Pco2) results in elevated intracranial pressure.12 Removal of fluid from the cisterna magna is contraindicated in a patient with increased intracranial pressure because a possibly fatal herniation of the brain through the foramen magnum may occur. Signs of increased CNS pressure include a moderate to marked decrease in mentation, mydriatic pupils, opisthotonos, extensor rigidity, ventrolateral strabismus, and papilledema.
inches) in cattle. In a cisterna magna tap, the needle entry site is close to the cervical spinal cord and the brainstem. To minimize the danger of central nervous system (CNS) damage during a cisterna magna tap, the animal should be adequately anesthetized and ventilated, because an increased partial pressure of carbon dioxide (Pco2) results in elevated intracranial pressure.12 Removal of fluid from the cisterna magna is contraindicated in a patient with increased intracranial pressure because a possibly fatal herniation of the brain through the foramen magnum may occur. Signs of increased CNS pressure include a moderate to marked decrease in mentation, mydriatic pupils, opisthotonos, extensor rigidity, ventrolateral strabismus, and papilledema.
Myelography
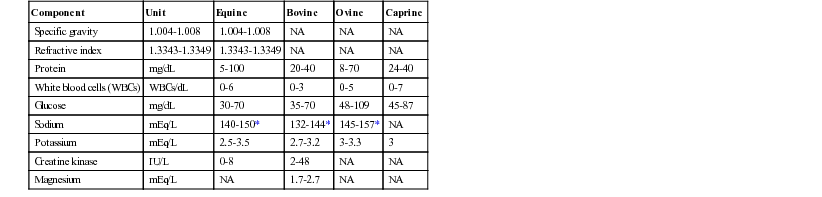
Analysis of Cerebrospinal Fluid
Component
Unit
Equine
Bovine
Ovine
Caprine
Specific gravity
1.004-1.008
1.004-1.008
NA
NA
NA
Refractive index
1.3343-1.3349
1.3343-1.3349
NA
NA
NA
Protein
mg/dL
5-100
20-40
8-70
24-40
White blood cells (WBCs)
WBCs/dL
0-6
0-3
0-5
0-7
Glucose
mg/dL
30-70
35-70
48-109
45-87
Sodium
mEq/L
140-150*
132-144*
145-157*
NA
Potassium
mEq/L
2.5-3.5
2.7-3.2
3-3.3
3
Creatine kinase
IU/L
0-8
2-48
NA
NA
Magnesium
mEq/L
NA
1.7-2.7
NA
NA
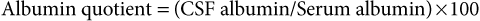
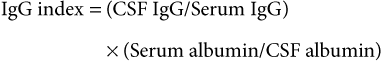
Diseases Presenting Principally with Forebrain Dysfunction or Multiple-Level Brain Dysfunction
Alphaviruses
History and Distribution
Western Equine Encephalomyelitis.
Eastern Equine Encephalomyelitis.
Venezuelan Equine Encephalitis.
Natural History
Eastern Equine Encephalomyelitis and Western Equine Encephalomyelitis.
Venezuelan Equine Encephalomyelitis.
Clinical Findings
Laboratory Findings
Diagnosis
Treatment
Public Health Significance
Postmortem Findings
Control
Miscellaneous and Foreign Emerging Viruses Causing Neurologic Signs
Getah/Ross River Viruses
Borna Disease
Henipavirus
California Serogroup Viruses
Equine Encephalosis
West Nile and Other Flavivirus Encephalitis
History and Distribution
Natural History
Clinical Signs

Diseases of the Nervous System
Chapter 35