Metabolic Control of Feed Intake
Implications for Metabolic Disease of Fresh Cows
Michael S. Allen, PhDa∗ and Paola Piantoni, Veterinaria, MSb, aDepartment of Animal Science, Michigan State University, 474 South Shaw Lane, Room 2265A, East Lansing, MI 48824, USA; bDepartment of Animal Science, Michigan State University, 474 South Shaw Lane, Room 2200, East Lansing, MI 48824, USA. E-mail address: allenm@msu.edu ∗Corresponding author.
Keywords
Hepatic lipidosis
Hepatic oxidation
Insulin resistance
Ketosis
Lipolysis
Metabolic disease
Propionic acid
Ruminal starch fermentability
Introduction
Suppression of appetite in dairy cows during the postpartum (PP) period results in negative energy balance (NEB), which increases the risk of metabolic disorders. Cows in the PP period are in a lipolytic state from low plasma insulin concentration and decreased insulin sensitivity of tissues, which results in a gradual increase in plasma nonesterified fatty acid (NEFA) concentration in the days before parturition. The liver extracts NEFA from the blood, and these can be esterified and stored as triglycerides (TG) or oxidized to acetyl CoA. Acetyl CoA is oxidized completely to CO2 or incompletely to ketones, which are then exported to the general circulation. Excessive storage of NEFA as TG compromises liver function while oxidation of NEFA might suppress feed intake. Satiety caused by hepatic oxidation of fuels might be a dominant mechanism controlling feed intake, especially in the peripartum period.1 Minimizing the rate of lipolysis and the duration of the lipolytic state are key to reducing incidence of metabolic disease; especially because feed intake is likely controlled by hepatic oxidation of NEFA. Lipolysis can be controlled by preventing excessive body condition, minimizing stress in the peripartum period, and maximizing energy intake in the PP period. Optimal body condition at parturition and increased energy intake PP can be achieved with strategic diet formulation. The objective of this article is to discuss metabolic control of feed intake in the peripartum period and its implications for metabolic disease of fresh cows. Understanding how feed intake is controlled during the transition from gestation to lactation is critical to both reduce risk and successfully treat many metabolic diseases.
Lipolytic state of cows in the peripartum period
The lipolytic state begins several weeks prepartum, with the decline in plasma insulin concentration and the decrease in insulin sensitivity of adipose tissue and muscle, and extends for up to several weeks PP. The decrease in insulin sensitivity of tissues is likely caused by the gradual increase in growth hormone,2 proinflammatory cytokines, such as tumor necrosis factor α3 from the placenta and adipose tissue, and increased plasma NEFA concentration.4 Signals related to the concomitant gestation initiate lipolysis to provide NEFA as a fuel to the liver and extrahepatic tissues, sparing the glucose required for fetal development. Euglycemia is maintained until initiation of lactogenesis,5,6 likely because glucose is spared by use of NEFA as a fuel. In addition, elevated NEFA concentration in blood is required to increase fat concentration of colostrum and milk in early lactation to meet the energy requirements of the calf. However, excessive fat mobilization can result in hepatic lipidosis, ketosis, and other metabolic disorders related to low feed intake and NEB. Importantly, oxidation of NEFA in the liver likely induces satiety, which might be the cause of the depression in feed intake.1 Suppressed feed intake following parturition prevents restoration of euglycemia, extending the lipolytic state for days or weeks PP. The depression in feed intake that occurs during the peripartum period is likely caused by the lipolytic state that is established several weeks prepartum and increases greatly in the days before parturition.
Hepatic export of ketones is beneficial to some extent because complete oxidation of NEFA might depress feed intake further. Therefore, in the PP period, a limited increase in ketone body concentrations in blood (plasma [BHB] <1.2 mmol/L7) should be considered normal, because lipid mobilization and NEFA oxidation is the expected response to decreased insulin sensitivity and plasma insulin concentration around parturition. Nevertheless, concentrations of BHB in plasma lower than 1.2 mmol/L are indicators of a good transition from gestation to lactation. In contrast, elevated plasma ketone body concentrations indicate a lipolytic state that likely suppresses feed intake. In this case, peripartum cows would benefit from a diet composition different from what might be optimal days or weeks later,1 when control of feed intake by hepatic oxidation subsides and signals from ruminal distention begin to dominate.
Metabolic diseases related to depressed feed intake
The depression in dry matter intake (DMI) PP results in a period of NEB for most cows, increasing the risk of metabolic disorders.8 The extent of lipid mobilization and, therefore, the depression in DMI, will determine if the cow will have normal plasma ketone body concentrations or will develop either subclinical or clinical ketosis. Incidence of ketosis is related to plasma concentrations of NEFA both prepartum9 and PP10 and to high body condition score (BCS) at parturition.11 Although ketosis might be secondary to other disease(s) that can depress DMI and increase NEB, it can also be primary, with no apparent cause. In both cases, feed intake suppression from hepatic oxidation of NEFA is probably a contributing factor.
The decline in DMI in the peripartum period is greater for cows with higher BCS,12 and this predisposes them to increased incidence of metabolic disorders, including ketosis and hepatic lipidosis. Cows with excessive body condition (BCS ≥3.5 on a 1 to 5 scale) during the transition period, likely have a greater rate and extent of lipolysis than cows with lower BCS because they have a larger mass of adipose tissue that can be mobilized and because the release of proinflammatory cytokines that contribute to insulin resistance13 is increased when the mass of adipose tissue is greater. Prolonged lipolysis is likely associated with a longer period of depressed feed intake and, therefore, NEB.
Liver uptake of NEFA is proportional to their concentration in the blood.14 Excessive hepatic TG concentration is a result of high blood NEFA concentration and compromises liver function, reducing gluconeogenesis15 and, therefore, restoration of blood glucose and insulin concentrations, further extending the lipolytic state. Because ruminant liver has limited capacity to export TG as very low density lipoproteins (VLDL),16 limiting lipolysis is the primary goal for a successful transition from gestation to lactation.
Other health disorders are associated with depressed feed intake around parturition. Depression in feed intake in the first several days PP greatly alters the balance between calcium absorbed and secreted in milk, increasing the risk of hypocalcemia. In addition, elevated blood NEFA concentration, and the resulting feed intake depression and increased ketones, will likely compromise the immune system response, increasing the risk of retained fetal membranes and metritis.17–19 Furthermore, depressed feed intake results in lower rumen digesta mass and volume increasing the risk of displaced abomasum from lower rumen fill and subacute ruminal acidosis from reduced buffer capacity of ruminal digesta.20
Control of feed intake
Feed intake control mechanisms are complex and involve multiple signals, redundancies, and levels of integration. Signals include distention from rumen fill (physical regulation), gut peptides (endocrine regulation), and oxidation of fuels (metabolic regulation). These signals are additive,21,22 and decisions to initiate and end meals are a result of integration of signals within brain feeding centers. The dominant signals controlling feeding behavior will likely vary temporally, within days, as well as through lactation.
Physical Regulation
Physical fill in the rumen has been studied extensively and previously reviewed.23 The filling effect of diets is determined primarily by forage neutral-detergent fiber (NDF), forage fragility (susceptibility to particle size breakdown by digestion and chewing), and digestion characteristics affecting turnover rate of fiber, such as the potentially digestible fraction and its rates of digestion and passage.24 Rumen distention is sensed by tension receptors located primarily in the reticulum and cranial sac of the rumen,25 conveyed to brain feeding centers, and integrated with other signals to determine feeding behavior. Signals from ruminal distention from physical fill likely dominate control of feed intake during the periods of highest milk production. This is in contrast to periods earlier and later in lactation, as discussed later in this article.
Endocrine Regulation
Gut peptides that potentially contribute to the control of feed intake include ghrelin, cholecystokinin (CCK), and the incretin glucagon like peptide 1 (GLP-1), which also may stimulate insulin secretion.26 Several of these have been studied in the ruminant animal27,28 but most of the work has been done in nonruminants because of their potential use as satiety factors in the treatment of obesity in humans.29,30 Ghrelin is known as the “hunger hormone” and is secreted by abomasal tissues. It is the only gut peptide known currently to stimulate feed intake, likely by increasing gastric emptying and passage rate from the rumen. Other gut peptides are secreted by the small intestine in response to various nutrients in the chyme. They may affect feed intake, gastric emptying, and favor digestion of nutrients through an increase in secretion of digestive enzymes or bile. A decrease in gastric emptying will likely result in longer retention times in the rumen and small intestine, favoring digestion and absorption of nutrients. Gut peptide signals (direct and indirect through effects on motility and distention as well as insulin and metabolism) are also integrated by brain feeding centers to determine feeding behavior.
Signal from hepatic oxidation of fuels
Research with laboratory species suggests that hepatic oxidation of fuels affects feeding behavior and that these signals are carried to brain feeding centers via hepatic vagal afferents; stimulation of oxidation inhibits feeding, whereas inhibition of fuel oxidation stimulates feeding. Various metabolic inhibitors have increased the discharge rate of hepatic vagal afferents and stimulated feeding, whereas hepatic vagotomy has blocked the stimulation of satiety by a variety of fuels in multiple species.31 This research indicates that fuel oxidation in peripheral tissues is involved in the control of feeding and that the transmission of signals to brain feeding centers is via hepatic vagal afferents. We call this the Hepatic Oxidation Theory (HOT) of control of feed intake1 and think it merits consideration for a better understanding of the prevention and treatment of metabolic diseases.
Energy Charge in the Liver
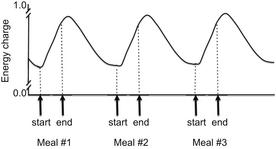
A series of experiments conducted by Friedman and colleagues32 showed that feeding behavior was related to hepatic energy status rather than fuel oxidation and ATP production per se. Energy status is measured by adenylate energy charge ([ATP] + 0.5[ADP])/([ATP] + [ADP] + [AMP]) and is related to phosphorylation potential. It is determined by the balance between the rate of production of high-energy phosphate bonds from oxidation of fuels and their rate of utilization by energy-consuming reactions. An increase in hepatic energy status has been related to decreased firing rate of hepatic vagal afferents and satiety. Firing rate increases in the period between meals as hepatic oxidation subsides and energy charge is depleted, resulting in hunger and meal initiation (Fig. 1). Even though we know the hypophagic response to fuel oxidation is likely linked to an increase in hepatic energy status, the mechanisms by which oxidation of fuels in hepatocytes affect the firing rate of the hepatic vagus are yet to be discovered.1
Fuels Oxidized in the Liver
Fuels extracted from the blood and oxidized in ruminant liver include NEFA, glycerol, lactate, and amino acids (mobilized from body reserves, produced by metabolism of other fuels, and provided by the diet), as well as propionate and butyrate, produced by microbial fermentation in the gastrointestinal tract. All these fuels have the potential to induce satiety at different degrees. Acetate and glucose are not extracted from the blood by the liver because the activity of the enzymes required for their activation and subsequent metabolism are low in ruminant liver.24 Consistent with HOT, their hypophagic effects in ruminants are much lower than fuels that are metabolized by ruminant liver.33
Effects of Propionate on Feed Intake
Hypophagic effects of propionate infusions (as propionic acid or sodium propionate) have been documented extensively for ruminants.24 Propionate is more hypophagic than acetate or butyrate when infused into the portal vein of sheep,34 and infusion of propionate into the mesenteric vein of steers reduced feed intake, whereas acetate infused at similar rates did not.35 Although propionate might be expected to decrease feed intake compared with acetate when infused at the same rate because it has a higher energy concentration, propionate linearly decreased metabolizable energy intake in lactating cows when infused intraruminally as iso-osmotic mixtures compared with acetate.36 The liver is likely involved in control of feed intake by propionate because hypophagic effects of portal infusions of propionate were eliminated by hepatic vagotomy.37 These studies suggest that hypophagic effects of propionate cannot be explained simply by the additional energy supplied compared with acetate. It is unlikely that animals consume feed to meet their energy requirements per se but rather have fuel-specific mechanisms regulating satiety and hunger.
Propionate is produced primarily by ruminal fermentation of starch. Of fuels metabolized by the ruminant liver, propionate is likely a primary satiety signal because its flux to the liver increases greatly during meals.38 Rate of propionate production and flux to the liver within meals (Fig. 2) is determined primarily by rate of eating, as well as the concentration and ruminal fermentability of dietary starch.24 Propionate is efficiently extracted by the liver and can be oxidized by conversion to acetyl CoA when its rate of extraction exceeds its rate of use for glucose production and acetyl CoA concentration is low. When cows are in a lipolytic state and hepatic acetyl CoA concentration is elevated, propionate oxidation is inhibited and propionate will be used as a glucose precursor. However, propionate entry into the tricarboxylic acid (TCA) cycle can stimulate oxidation of the existing pool of acetyl CoA (Fig. 3),1 generating ATP and increasing energy charge. Control of feed intake by propionate in the PP period as well as implications for diet formulation will be discussed in later sections.
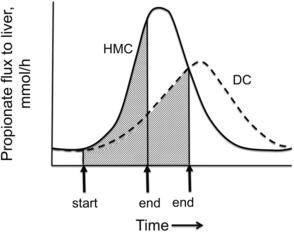
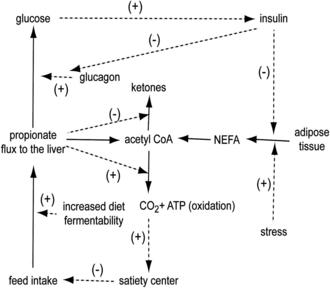
Control of feed intake in the PP period
The “flood” of readily oxidizable NEFA to the liver is likely responsible for the depression of feed intake during the peripartum period consistent with the inverse temporal relationship between plasma NEFA concentration and DMI observed in many studies.1 The following sections detail hepatic oxidation of fuels to provide a better understanding of how diet likely interacts with lipolytic state to affect energy intake.
Oxidation of NEFA in the Liver
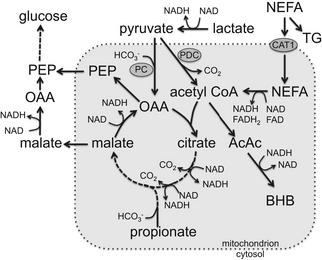
Carnitine acyl transferase (CAT1) transports NEFA into mitochondria by binding them to carnitine. The balance between transport of NEFA into mitochondria and their esterification and storage as TG is affected by several factors, including availability of carnitine and concentration and activity of enzymes. Supplementation of carnitine to transition cows increased in vitro oxidation of palmitate39 and depressed feed intake in the early PP period,40 consistent with HOT.1 Moreover, CAT1 is inhibited by malonyl CoA (an intermediate in fatty acid synthesis) and methylmalonyl CoA (an intermediate in propionate metabolism). Once inside the mitochondrion, reducing equivalents (FADH2 and NADH) are generated by β-oxidation of NEFA. These reducing equivalents can then generate ATP, thus increasing the energy charge of the cell. It is important to recognize, however, that reducing equivalents must undergo oxidative phosphorylation (OXPHOS) to generate ATP. Therefore, there may be a lag phase during which reducing equivalents may accumulate before ATP generation, thus they may not immediately affect energy charge, as discussed later.
Each cycle of β-oxidation shortens the FA by 2 carbons, yielding 1 molecule of acetyl CoA, FADH2, and NADH. The process continues until the entire chain is cleaved into acetyl CoA units (or until there is a propionyl CoA terminal in the case of odd-chain FA). Acetyl CoA, in turn, is either oxidized in the TCA cycle or exported as ketone bodies (Fig. 4). Each cycle of β-oxidation yields at least 14 ATP if acetyl CoA is oxidized in the TCA cycle, but only 4 if it is exported as ketone bodies. If feed intake is controlled by hepatic energy status, exporting acetyl CoA from the liver as ketone bodies is expected to help alleviate the depression of intake, because ketogenesis would not generate as great an increase in energy charge as would complete oxidation of acetyl CoA.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


