Chapter 44 Chemotherapy
Principles of Chemotherapy
Gompertzian growth kinetics have been used to describe tumor growth.1 In the Gompertzian model, the tumor growth fraction increases exponentially over time. Once the limit of clinical detection is reached, tumor growth progressively slows down and eventually plateaus. Response to chemotherapy highly depends on the location of the tumor on its growth curve. For example, tumor cells in the latent growth phase (i.e., prior to clinical detection) are more sensitive to chemotherapy than cells in subsequent phases because the growth fraction is greater during latency. In general, smaller tumors have higher growth fractions compared with larger tumors; consequently, they are more susceptible to chemotherapy.
Several factors influence the ability of chemotherapeutic agents to kill cancer cells. These include (a) intrinsic or acquired resistance to a chemotherapeutic agent; (b) impaired drug delivery across cell membranes; (c) duration of exposure to an effective concentration of the drug; (d) upregulation of glutathione, glutathione-S-transferases, or other detoxification pathways; (e) dysregulation of apoptotic (cellular death) pathways; or (f) activation of survival pathways by growth factors, such as epidermal growth factor. Acquired resistance to chemotherapy is often associated with overexpression of P-glycoprotein, a transmembrane pump encoded by the multiple-drug resistance gene.2
It is unclear as to whether any tumor cell or only stem cells develop resistance. Tumors develop intrinsically resistant cell lines (or clones) because of genetic instability that cause a susceptible tumor to become subsequently unresponsive to chemotherapy and to relapse.3 Based on this theory, induction chemotherapy should consist of a combination of drugs rather than a single agent.4
Resistance can also be explained by the “tumor stem cell theory,” according to which committed cancer cells are killed by induction chemotherapy whereas stem cells survive and repopulate the tumor.5
Chemotherapeutic agents are generally administered at the maximum tolerated dose and at the highest dose intensity (i.e., shortest dosing interval). In addition, they are commonly dosed based on the estimated body surface area. As a result of the increased toxicity observed with this dosing scheme in cats and small dogs receiving drugs such as doxorubicin and carboplatin, it is currently recommended, that the dose of specific chemotherapeutic agents be calculated on an mg/kg basis for animals weighing less than 10 kg rather than on the estimated body surface area.6 The author applies the mg/kg dosing scheme to dogs weighing less than 15 kg.
Classes of Chemotherapeutic Agents
Alkylating Agents
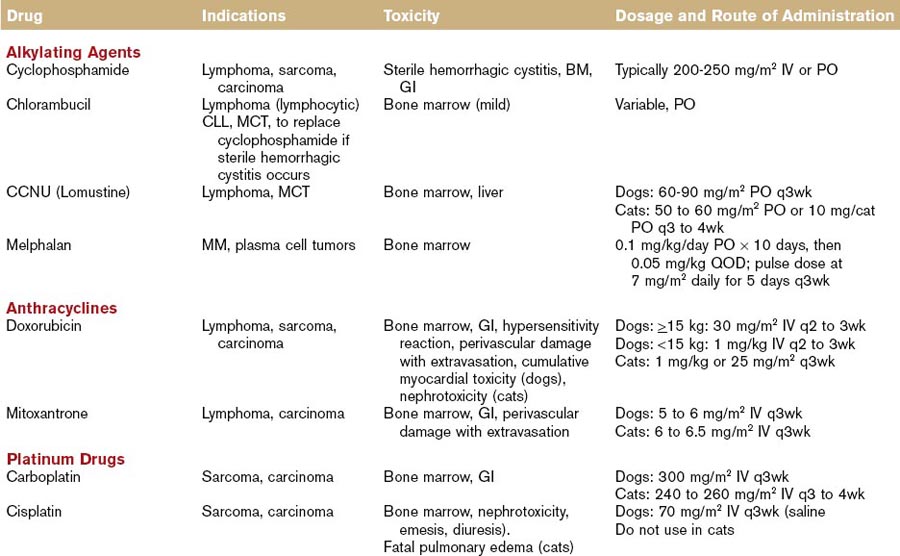
The first nonhormonal chemical found to have antitumor properties was a nitrogen mustard alkylating agent (sulfur mustard gas). When used as a weapon during World War II for its vesicant effects, the gas resulted in bone marrow suppression among the victims. This led to the development of a nitrogen mustard compound (mechlorethamine or Mustargen) and to its clinical application for the treatment of various cancers, primarily lymphomas.7 Alkylating agents react with DNA strands by inserting an alkyl group (thus the term alkylating), and changing the DNA structure. Some agents are monofunctional (i.e., they react with only one strand of DNA), some are bifunctional (i.e., they react with both DNA strands to produce a “crosslink”), and they are cell-cycle nonspecific. The most commonly utilized alkylating agents in veterinary oncology are chlorambucil (Leukeran), cyclophosphamide (Cytoxan), mechlorethamine (Mustargen), melphalan, and lomustine (Table 44-1). A unique toxicity to cyclophosphamide is the possible development of sterile hemorrhagic cystitis, an irritation of the urinary bladder caused by the inactive metabolite acrolein.
Anthracyclines
Anthracycline antibiotics are derived from the bacteria Streptomyces peucetius. Doxorubicin, a hydroxylated daunorubicin derivative, is widely utilized in human and animal patients for a variety of malignancies, including lymphoma, sarcomas, and carcinomas. It is cell-cycle nonspecific and exerts its cytotoxic effect through different mechanisms, including free radical formation, DNA intercalation, and inhibition of protein synthesis. It also inhibits topoisomerase, leading to the formation of cleavable complexes, DNA damage, and cellular death. It is metabolized by the liver and eliminated primarily in the feces. Doxorubicin can promote oxidative reactions, forming highly reactive oxidative species, including superoxide anions, hydrogen peroxide, and hydroxyl free radicals that can damage lipid membranes. Cellular defenses against free radical formation (e.g., catalase, glutathione) become saturated and because catalase levels are low in cardiac muscle of humans and dogs, cardiotoxicity is a potential side effect of this drug. Nephrotoxicity has been observed in cats treated with doxorubicin, but the pathogenesis is unclear.8
Mitoxantrone is a synthetic antitumor antibiotic that was originally developed as a noncardiotoxic alternative to anthracycline compounds. It inhibits topoisomerase IIα by stabilizing topoisomerase IIα–cleavable complexes. Unlike doxorubicin, mitoxantrone does not result in free radical formation, which may account for its reduced cardiotoxic potential. Mitoxantrone is metabolized in the liver and is eliminated via feces and urine. In humans, it is used for the treatment of non-Hodgkin lymphoma and acute leukemias, as well as for the treatment of breast cancer and hormone-refractory prostate cancer (see Table 44-1).9
Platinum Drugs
The antitumor activity of platinum coordination complexes was first recognized by Rosenberg and colleagues in 1961. They observed that cisplatin induced filamentous growth in bacteria without affecting RNA or protein synthesis (suggesting DNA as the primary target of the drug).10 Platinum drugs act preferentially at the N7 position of guanine and adenine residues, forming mono- and bifunctional adducts. Initially, monofunctional adducts form and these may subsequently form intra- or interstrand crosslinks. The toxicity profile of cisplatin, as observed in early clinical trials, led to the development of platinum analogues (carboplatin, oxaliplatin) with less toxicity and potentially greater efficacy against various tumor types. They are cell-cycle nonspecific, and elimination occurs through the kidneys (see Table 44-1).
Antimetabolites
5-Fluorouracil
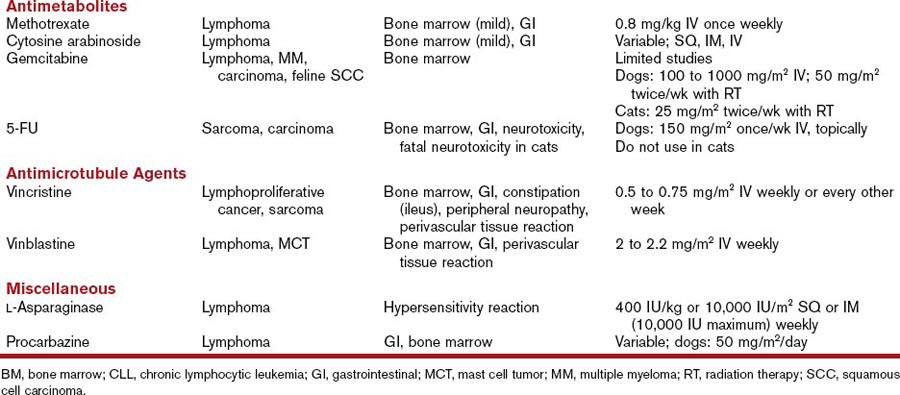
5-Flourouracil (5-FU) is a fluoropyrimidine that requires intracellular activation to exert cytotoxicity. It enters the cells through a facilitated uracil transport mechanism and is subsequently converted into the active metabolite 5-fluoro-2′-deoxyuridine monophosphate (FdUMP), which interferes with DNA synthesis and repair. Elimination occurs through the lungs and the kidneys (see Table 44-1).
Antimicrotubule Agents
The vinca alkaloids and paclitaxel are metabolized by the liver and excreted in the feces (see Table 44-1).11
Miscellaneous Agents
Imatinib mesylate (Gleevec) is a tyrosine-kinase inhibitor that occupies the adenosine triphosphate (ATP) binding site of the Bcr-Abl tyrosine kinase and of other tyrosine kinases, platelet-derived growth factor, c-kit, and stem cell factor. In humans, imatinib is considered the first-line therapy for chronic myeloid leukemia and for gastrointestinal stromal tumors (GISTs) expressing the c-kit tyrosine kinase (see Table 44-1).12
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


