Chapter 5 Cellular Growth/Neoplasia
The balance between cell growth and death is essential in maintaining tissue and organ homeostasis, even in simple biologic systems. This is of particular importance in specialized organs of high cell turnover such as the gastrointestinal (GI) tract of larger mammals. Several mechanisms of control have evolved within these organs to maintain this balance, including regulators of cell-cycle progression, proliferation, and death. These regulators are influenced by both local (paracrine and autocrine) and systemic (endocrine) signals.1 Derangements in any of these control mechanisms can result in a disruption of the balance between proliferation and death, often leading to abnormal proliferation and potentially to cancer. The effectiveness of the control mechanisms is evidenced by the relatively infrequent development of cancer of the GI tract despite the extraordinary and diverse requirements for cellular turnover and differentiation of this tissue. This chapter reviews the pathways and processes that control cell growth and proliferation, with specific focus on those that when dysregulated result in cancer of the GI tract.
Overview of Normal Cell Growth and Proliferation
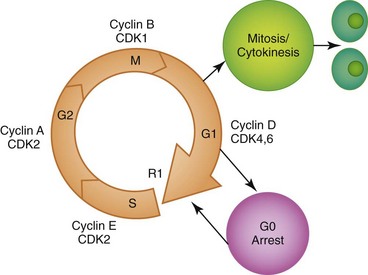
For most cell types, regulation of cell division and replication is a highly conserved continuum with cells traversing through the stages of interphase, mitosis, and cytokinesis (Fig. 5-1). Interphase is defined as the period prior to cell division where the cell grows and replicates its chromosomes in preparation for division by mitosis. For somatic cells, cellular replication is accomplished during mitosis and cytokinesis. The result of mitosis is the generation of two daughter cells (cytokinesis) each with a diploid (2n) genetic constitution. For germline cells that are committed to cellular reproduction, cells exit interphase and enter meiosis. In meiosis, chromosomal numbers within a nucleus are halved (1n) after two cell divisions. Following cytokinesis, two haploid germ cells are available for reproduction.2
Interphase
During interphase, cells undergo DNA duplication and a period of extensive RNA, protein, and lipid synthesis. This stage is composed of three signal dependent phases: growth-1 (G1), synthesis (S), and growth-2 (G2), that is then followed by mitosis (M) (see Fig. 5-1). Cells that have reached terminal differentiation or the nonreplicating phase, exit from the cycle toward the end of the G1 phase and arrest in a phase called G0. Several mechanisms control cell growth and proliferation during the transitions between G1 and S and between G2 and M in the cell cycle (also termed checkpoints). The key players that are involved in the regulation of these transitions are cyclins and cyclin-dependent kinases (CDKs).3
Cyclins and Cyclin-Dependent Kinases
The entry of cells from the phase of mitosis to G1 involves the presence of progrowth external factors and appropriate internal mitogenic signals. Under circumstances that favor proliferation, the cell cycle progresses as CDKs associate with small intracellular regulatory subunits called cyclins. Cyclins, unlike CDK proteins, have variable expression patterns depending on the cell-cycle phase, acting as a primary level of control for CDK activation. For instance, expression of cyclins A and B is highest at the S/G2 phase transition, whereas expression of cyclins D and E are highest at G1/S.4
Prior to becoming functional, the cyclin–CDK complexes must become phosphorylated by cyclin-activating kinases (CAKs). This process is site-dependent, meaning that phosphorylation may result in either suppression or activation of the complex, depending on the specific phosphorylated residue.5 Once activated, the cyclin–CDK complexes can then exert their regulatory control via the phosphorylation of target proteins.
During G1, cyclin D activates either CDK4 or CDK6, resulting in a functional cyclin–CDK complex. When activated, this complex results in the phosphorylation of several target proteins, including the retinoblastoma protein (Rb).6 Once the Rb is phosphorylated, the transcription factor E2F is liberated and enables not only the transcription of a variety of genes but also the induction of cyclin E. Cyclin E, in association with CDK2, maintains the Rb in a phosphorylated state. It is the phosphorylation of the Rb that is considered the quintessential final guardian of the transition from G1 to S phase, and is the critical point at which a cell will move into S phase or into G0, otherwise known as the restriction point. Elevated levels of phosphorylated Rb allow for the accumulation of free E2F, which increases the level of cyclin A and enables the cell to progress into DNA synthesis, or S phase. Cyclin A then plays a critical role in S phase as it interacts with CDK2 and CDK1 while cyclins D and E are concurrently degraded. CDK2 and CDK1 are key for the transition through S phase into M phase, or mitosis. In particular, CDK1, in association with cyclins A and B, is required for the phosphorylation of cytoskeletal proteins necessary for mitosis.3,7
In addition to the cell-cycle dependent expression of cyclins, and the regulatory control of the CAKs, the cell cycle is also regulated by the activity of antagonistic cyclin-dependent kinase inhibitors (CDKIs). These proteins block progression of the cell from G1 to S phase by binding cyclin–CDK complexes and blocking the activation of downstream proteins. Three types of CDKIs are recognized and include the INK4 family, CIP, and KIP.8 The INK4 family of CDKIs exhibits high specificity for CDK4 and CDK6 complexes, preventing the association of these proteins with cyclin D. CIP (WAF1/p21) is regarded as a general CDK inhibitor, whereas KIP (p27) functions via activation of transforming growth factor (TGF)-β that subsequently prevents the activation of cyclin E/CDK2 and arrests the cell in G1.9
Ligands and Transmembrane Receptors
The internal regulation of the cell cycle is modulated by external and extracellular environmental cues. There are four recognized pathways that play a part in conveying these extracellular messages into the cell: tyrosine phosphorylation; serine and threonine phosphorylation; generation of signaling nucleotides via G-protein-coupled receptors; and production of calcium phosphoinositol through stimulation of phospholipase C.10 Signaling through these pathways provides a connection between the cell cycle and the environment in which a cell resides. In the presence of growth factors, cells may more easily transition between cellular checkpoints, and in some cases will by influenced toward cellular survival rather than death (discussed below in “Overview of Cellular Death and Apoptosis: The Response to DNA Damage”).11
Mitosis and Cytokinesis
Once a cell has committed to replicate and surpasses the checkpoints at G1/S and then G2/M, it enters mitosis. Mitosis is characterized by cellular DNA replication and segregation, organized in four different phases.2 Prophase, the first phase of mitosis, involves the replication of chromosomes (4n) and the division of chromosomes into two sister chromatids. These chromatids are joined by a central centromere. In the cytoplasm, centrioles and asters develop and then align at either pole of the nucleus. The nuclear membrane disintegrates at the end of prophase while the centromeres form spindle fibers that will become the organizational scaffolds for chromosomal assembly. In metaphase, the chromosomes are aligned via centriole-spindle fiber interactions on the middle of the spindle, or the metaphase plate. Once aligned, the centromeres split and individual chromatids are segregated to either centriole by the regressing spindle fibers (anaphase). The final phase of mitosis, or telophase, involves the reappearance of the nuclear envelope around each centriole and its associated chromatids. The shared cytoplasm between these two new nuclei is then split (cytokinesis). The maintenance of DNA ploidy, or normal diploid genotype, is critical in mitosis. Aneuploidy, or aberrations in the normal 2n chromosomal number, often resulting from dysregulated mitosis, is a frequent change noted in cancer.10,12
Overview of Cellular Death and Apoptosis: The Response to DNA Damage
Exposure to cellular stressors, hypoxia, and toxins can result in damage or changes in the genetic material (DNA) of a cell. In a preserved checks-and-balances system, these changes are identified by a cell and result in its arrest within the cell cycle, most often at a cellular checkpoint. During cellular arrest the damaged DNA is surveyed and repaired. If the damage is determined to be extreme or nonrepairable the cell undergoes controlled cell death (apoptosis). This process of arrest, survey, repair, or death ensures high-quality, mutation-free replication needed for optimal organ homeostasis. Cell-cycle control genes function at each of the cell-cycle cellular checkpoints in a nonmutually exclusive manner. The best-characterized checkpoint gene is the tumor-suppressor gene p53 (Fig. 5-2).13 The activation of p53 relies upon phosphorylation by either DNA-dependent protein kinase or by ATM kinase in response to cellular damage. Once phosphorylated, p53 functions as a transcriptional activator of multiple cell-cycle regulator genes, including p21 (CIP1/WAF1).14,15 p53 mediates cell death in part by downregulating the expression of the oncogene bcl2, an inhibitor of apoptosis, which shifts the balance toward cell death. Accumulations in proapoptotic signals, such as bax, tissue necrosis factor, Fas ligand, and the activation of other oncogenes, also promote cellular apoptosis through p53 regulation.
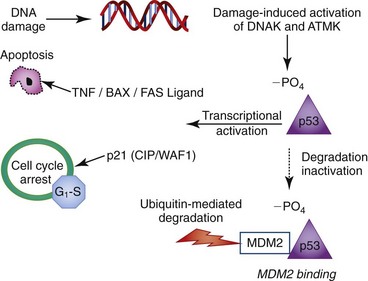
p53 is also modulated by a variety of proteins to ensure proper function. The gene MDM2 (mouse double minute 2 oncogene) functions as an antagonist to p53.16 MDM2 can target p53 for degradation by binding the protein and tagging for proteosome-mediated degradation. Conversely, p53 also has a defined binding site in the MDM2 internal promoter that competitively binds free p53. Significant MDM2-p53 binding results in decreased circulating p53, suppressing p53 transcriptional activity.17 The collective result is that the p53 available for phosphorylation and subsequent transcription of proapoptotic/proarrest proteins drops, favoring cell survival.
The “Cancer Genes”: Oncogenes and Tumor-Suppressor Genes
When mutated, certain genes confer malignant characteristics across multiple tumor types. Many of these “cancer genes” have functions related to control of the cell cycle or apoptosis. Two broad categories of cancer genes are known to exist. Oncogenes represent normal genes (protooncogenes) that undergo “gain-of-function” in cancer that contribute to cell proliferation or reduced cell death. Conversely tumor-suppressor genes normally restrain cell proliferation or promote appropriate cell death and undergo “loss-of-function” in cancer.18
Oncogenes
Studies of RNA tumor viruses (retroviruses) provided the first evidence that genetic factors play a role in the development of cancer. Rous demonstrated that a retrovirus, now known as avian leukosis virus, was capable of producing lymphoid tumors in chickens.19 Retroviral sequences that are responsible for transforming properties are called viral oncogenes (v-onc). The names of these genes are derived from the tumors in which they were first described (e.g., v-ras from rat sarcoma virus). Subsequently, viral oncogenes were shown to have cellular homologues called cellular oncogenes (c-onc).20 The term protooncogene was later used to describe cellular oncogenes that do not have transforming potential to form tumors in their native state but which can be altered to lead to malignancy. Most protooncogenes are complex genes involved in the control of cell growth and proliferation. These genes, when transcribed, represent a variety of proteins, including signaling growth factors, growth factor receptors, protein kinases, signal transducers, and nuclear proteins and transcription factors. Abnormal function/expression of any of these proteins can result in changes that lead to aberrant proliferation.
Growth factors act through transmembrane receptors on the cell surface and trigger the intracellular cascades that ultimately result in cellular proliferation. Overexpression of a growth factor and the abnormal expression of a growth factor by a cell that typically does not produce it represent two ways in which a cellular environment can contribute toward carcinogenesis. Examples of growth factors that are typically expressed by tumor cells are epidermal growth factor (EGF) and fibroblast growth factor (FGF).21
Signal transduction through the cell involves a variety of second messengers, such as guanosine triphosphate (GTP), adenosine triphosphate (ATP), and intracellular calcium. If mutated, the genes that transcribe these proteins may alter the response of these second messengers to inhibitory or activation signals. A common signal transducer that is altered in malignancies is GTP. GTP is converted to guanosine diphosphate (GDP) via the guanosine triphosphatase (GTPase) activity of G proteins as part of the signaling cascade. This cascade is modulated by the expression of the ras family of genes, where the resultant proteins police GTPase and GTP binding activity. When mutated, the ras oncogene allows for continual expression of GTP, reduced numbers of the CDKI p21 (CIP), and reduced GTPase activity. These changes together favor tumor formation.22
Alterations in the level and activity of intranuclear transcription factors may allow for aberrant gene expression and induce a malignant phenotype. An example of a key nuclear oncogene is that of the myc family and in particular c-myc. c-myc plays a significant role in modulating cellular proliferation, differentiation, and apoptosis amongst other functions. Mutations in c-myc are frequently encountered across tumor types, including those in the GI tract.23
In general, oncogenes require mutation of only one allele in order for phenotypic change to occur. This is known as a dominant gain-of-function. The conversion from protooncogene, the normal phenotype, to oncogene, the mutated form, can involve a variety of mechanisms, commonly relying upon abnormal gene amplification. Other mechanisms of mutation include chromosomal translocation, point/frameshift mutations, and viral insertions.24
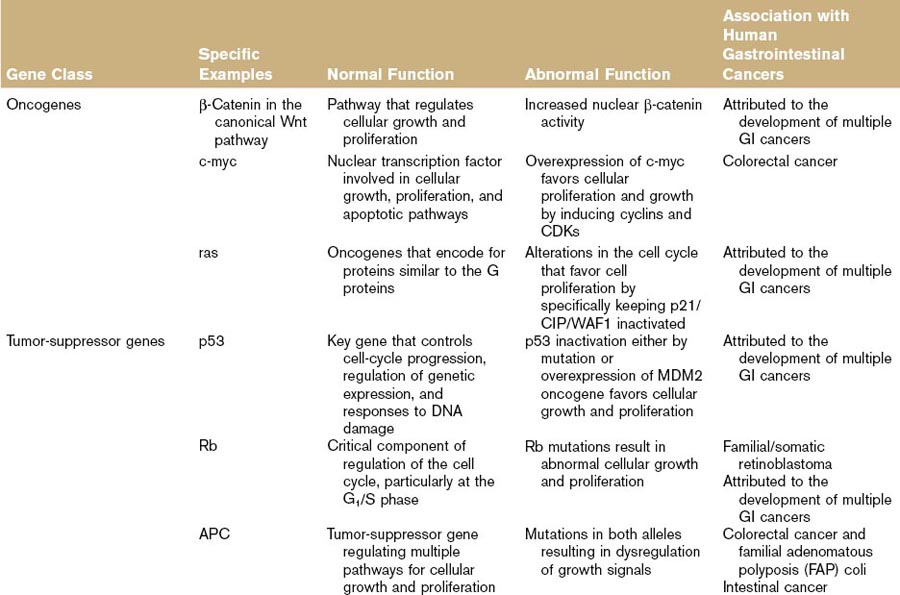
Table 5-1 lists the oncogenes associated with the development of GI cancer. The oncogene β-catenin (and alternations in the Wnt [wingless]–β-catenin pathway) has been specifically incriminated in the development of GI cancers. This pathway involves the regulation of cytoplasmic free β-catenin and conformational changes in the APC–axin–GSK-3B complex. Nuclear translocation of this β-catenin and subsequent interactions with nuclear proteins result in the active transcription of a variety of genes including c-myc, cyclin D1, MMP-7, and ITF-2, most of which are progrowth proteins that have been implicated in cancer development.25
Tumor-Suppressor Genes
Tumor-suppressor genes differ from oncogenes in that both alleles encoding a gene must be converted in order for a phenotype to be seen. Maintenance of a single allele is sufficient for protein expression to maintain cellular phenotype. When tumor-suppressor genes are mutated, the result is a loss of inhibitory function and the promotion of cellular growth and proliferation.26 Two common tumor-suppressor genes that have been studied extensively and implicated in a variety of human and animal tumors include the Rb and the p53 tumor-suppressor genes.
As illustrated previously, the Rb plays a crucial role in the decision of a cell to proceed from G1 to S phase.6 Disruption of the Rb or other proteins that work in concert with the Rb can lead to unwanted cellular proliferation. Observations of the familial inheritance patterns of certain cancers, like that of the inherited form of retinoblastoma led to the formation of Knudson’s two-hit model. This hypothesis suggests that two separate mutations must occur in a tumor-suppressor gene prior to tumor development. Mutations or dysregulation in the Rb pathway are common in many different forms of cancer in both man and animals.27–33 The inherited form of retinoblastoma results from a germline mutation in one allele of a gene that exists in all cells that derive from that altered cell. This loss is referred to as the loss of heterozygosity or allelic deletion. A secondary somatic mutation (second hit) occurs in the other normal allele of that same gene, resulting in the so-called two-hit knockout of tumor-suppressor function.34 In the sporadic form of retinoblastoma, two distinct somatic mutations are needed in both the Rb alleles to eliminate tumor-suppressor function and lead to cancer development. Not surprisingly, the sporadic form of retinoblastoma occurs in older patients and is less commonly bilateral at presentation.
The tumor-suppressor gene p53, sometimes known as the “guardian of the genome,” controls cellular arrest, apoptosis, and proliferation, and is essential in mitigating DNA damage. The policing activity of p53 prevents the accumulation of mutations that would favor genomic instability and subsequent malignancy.14 Inactivation of p53 by mutation plays a critical role in tumorigenesis in many human and animal cancers.35–39 For the most part, dysregulation of p53 protein is associated with its stabilization and overexpression in cancer tissues. Germline mutations that affect p53 also have been documented and associated with familial cancer risk (Li-Fraumeni syndrome).40 The activity of p53 can also be affected by nonmutational means. Overexpression of the p53 inhibitor MDM2, a cellular oncogene, reduces the amount of active p53, shifting the cell balance away from apoptosis.17
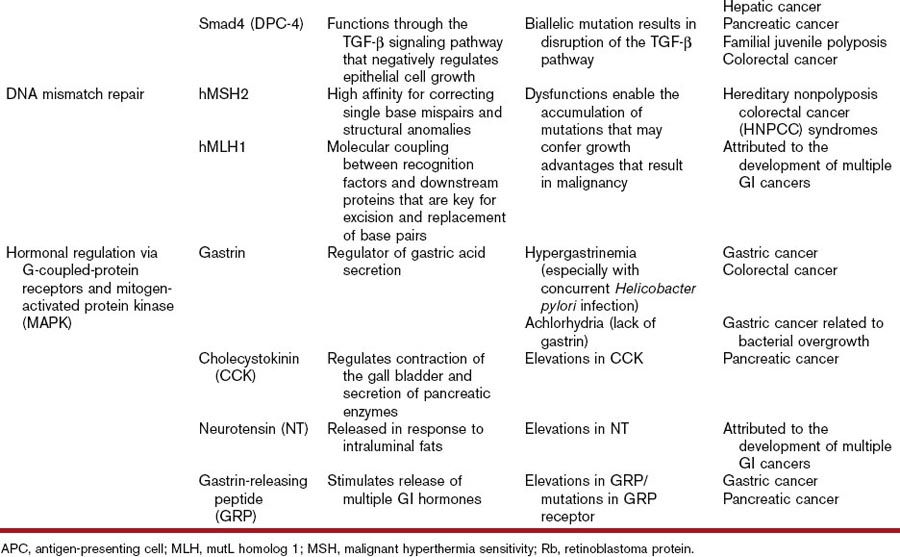
Another key tumor-suppressor gene linked to the development of several GI neoplasms is Smad4 (DPC4). Smad4, a member of the Smad family, plays a critical role in the mediation of the TGF-β pathway by acting as signal transducer. Wild-type expression of this gene results in suppression of epithelial cell growth via regulation of transcription of target genes inside the nucleus.10 Germline mutations in Smad4 have been implicated in the development of familial juvenile polyposis, a precancerous condition of the GI tract. Somatic mutations in this same tumor-suppressor gene have also been linked to the development of pancreatic and colorectal cancers.41
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


