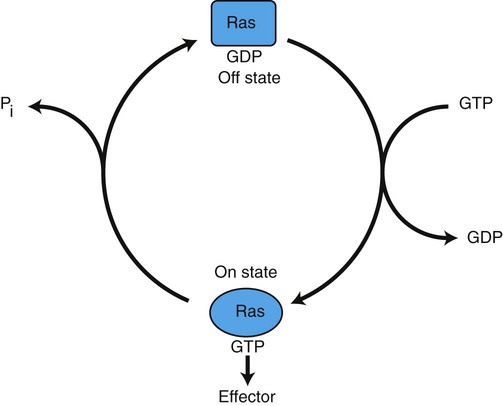
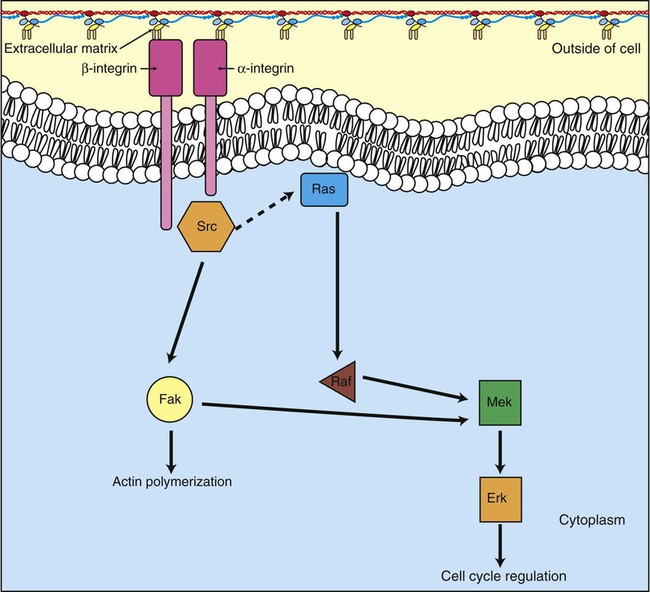
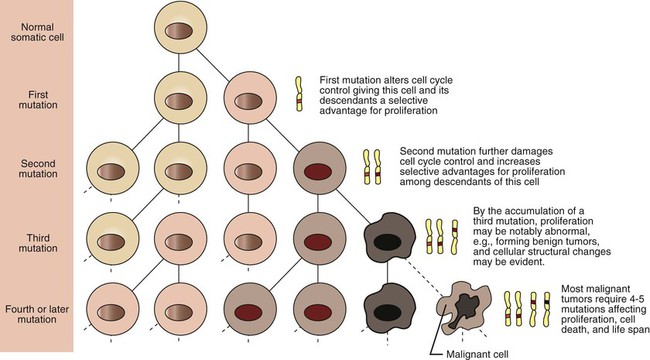
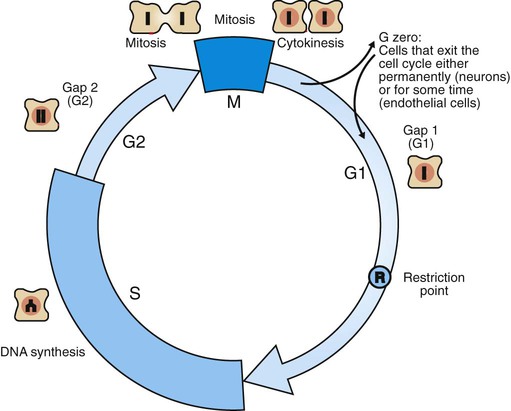
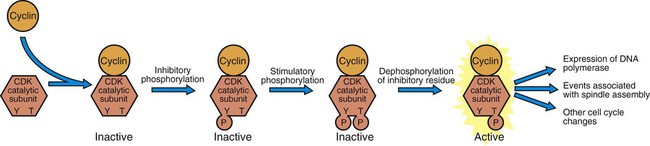
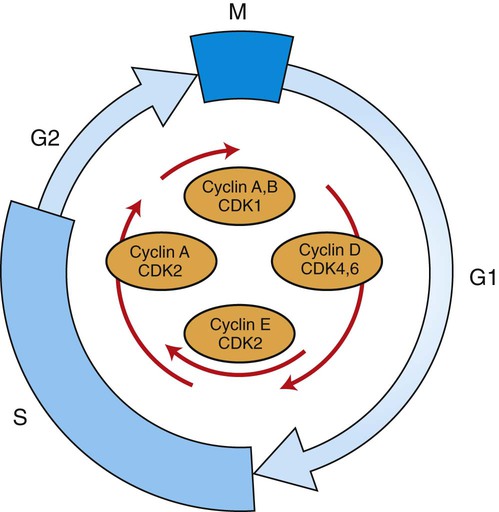
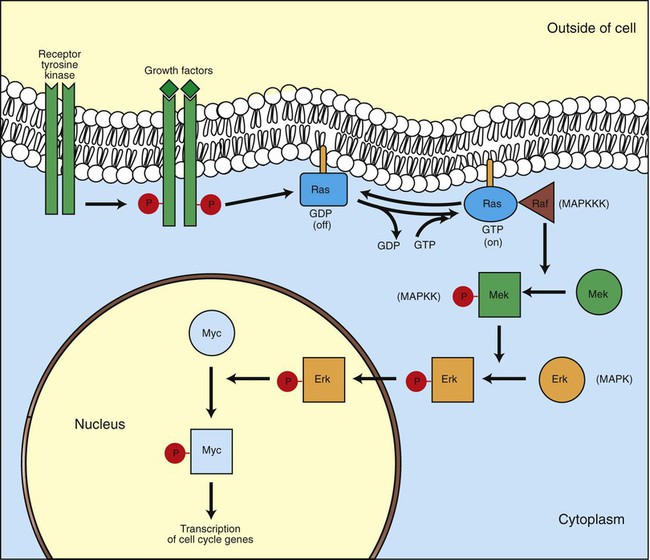
1. Cancer arises from genetic dysfunction in the regulation of the cell cycle, cell life span, and cell suicide. Control of the cell cycle (proliferation) 1. Cell division is the result of a clocklike cell cycle. 2. Cyclin-dependent kinases are the “engines” driving the cell cycle. 3. The CDK “engines” are controlled by both throttle (oncogene) and brake (tumor suppressor) controls. Growth factor pathway: principal stimulator of cell proliferation 1. The cell cycle is stimulated by growth factors that bind to and activate receptor tyrosine kinases. 2. The ras oncogene contributes to many cancers and serves as a model for understanding small G proteins. 3. The MAP kinase pathway leads to the expression of cyclins and other stimulators of the cell cycle. 4. The MAP kinase pathway also mediates the stimulation of the cell cycle by cell adhesion. Tumor suppressors: inhibitors of cell cycle 1. Checkpoints in the cell cycle are manned by tumor suppressors. 2. The retinoblastoma and P53 proteins are the main gatekeepers for the cell cycle. Mechanisms regulating cell suicide and cell life span 1. Apoptosis is the process of cell suicide. 2. Resistance to apoptosis via the intrinsic pathway is a hallmark of cancer. 3. Cellular life span is determined by DNA sequences at the ends of chromosomes. Tumor origin and the spread of cancer 1. Cancer cells may be related to stem cells. 2. Death by cancer is usually the result of its spread, not the original tumor. 3. Growth of solid tumors depends on development of new blood vessels. Traditionally, cancer was (and often still is) first detected in humans and domestic animals by clinicians feeling for an unusual mass of cells, tumor cells. Thus, cancer is quite intuitively a disease affecting cellular growth. In the last 25 years, enormous progress has been made in understanding several normal control pathways that regulate cell growth, as well as how these Rube Goldberg pathways (see Chapter 1) go wrong in cancer. Cancer is a genetic disease (but not usually a hereditary disease) and a uniquely cellular disease. As shown in Figure 2-1, tumors and other cancers arise from the division of a single mutant cell whose descendants accumulate several additional mutations to become increasingly damaged with respect to control of cellular proliferation, life span, and cell death. That is, cancer is a genetic disease caused by the accumulation of mutations in body cells, such as those of the epithelia lining the lungs or the secretory epithelia of the mammary glands. All the cells of a tumor can trace their ancestry back to a single cell that developed an initial deleterious mutation. This first mutation usually occurs in a gene controlling proliferation, such that the cell produces a mutant protein1 that is a dysfunctional, more permissive regulator of the cell cycle. This greater “permissiveness” provides the mutant cell with more opportunity to proliferate, and it thus has a selective advantage compared with its normal neighbors. Perhaps because of this selective advantage, or because of continued exposure to mutagens (e.g., cigarette smoke, agricultural chemicals), a descendant of this cell accumulates another mutation that also affects some aspect of the cell cycle or cell death. This increases the doubly mutant cell’s selective advantage further still, and the downward spiral of increasingly abnormal, dividing cells begins to spin out of control. Scientists agree that this accumulation of mutations in individual genes is necessary for cancer to develop, but some think it is not sufficient. Rather, they argue that cancer only results when the accumulation of mutations eventually leads to large-scale genetic instability, such that whole chromosomes are gained and lost. The majority of spontaneous tumors do have cells with abnormal sets of chromosomes, a phenomenon called aneuploidy. Whether aneuploidy is necessary for cancer remains to be seen, but there is no disagreement that cancer cells are in some way badly damaged with respect to genes controlling growth. The Rube Goldberg device that controls cell growth is particularly complex, with many, many more components than the “garage door opener” of Figure 1-13. To explain these pathways, we begin with the cell cycle that, like the carriage house door, is near the end of the system of control. That is, most of the control elements feed “downstream” to control the cell cycle, or intersect with some aspect of cell cycle control. Figure 2-2 shows the classic diagram of the cell cycle in which the cell changes its state toward division, progressively going around the diagram, like the hands of a clock. For most mammalian cells, the duration of one cell cycle in culture varies between 18 and 30 hours. Two phases of the cell cycle were identified first and seemed to be where the most important events of the cell cycle occurred. One is synthesis (S) phase, during which the DNA is duplicated. The second is mitosis (M) phase, during which the duplicated chromosomes are separated to opposite sides of the cell and the cytoplasm divides. In addition to the obvious need for such events if cells are to reproduce, note that both phases must be highly precise. It is crucial for the cell that DNA synthesis produces exactly twice the original amount of DNA, no more and no less. Otherwise, there will not be two identical copies of the genetic material to pass on to two identical cells. Similarly, the machinery segregating the duplicated chromosomes during mitosis must partition exactly equal numbers and types of chromosomes to daughter cells, or the cells will be aneuploid. If DNA is not precisely replicated, or if the chromosomes are not properly aligned, the cell cycle is halted, by checkpoints, as described later. Recall from Chapter 1 that protein kinases, which are enzymes that phosphorylate other proteins, are important as elements of signaling pathways. For example, the second messenger cyclic adenosine monophosphate (cAMP) acts by activating protein kinase A (see Figure 1-18), and diacylglycerol as a second messenger activates protein kinase C (see Figure 1-19). Protein kinases play a major role in many aspects of control of the cell cycle; most importantly, CDKs, when activated, can directly cause a cell to enter either S phase or mitosis, whether the cell is ready or not. Active CDKs are composed of two different types of protein subunits (Figure 2-3). The catalytic subunits (numbered CDK1, CDK2, etc.) are the subunits that have enzymatic activity for hydrolyzing adenosine triphosphate (ATP) and transferring the phosphate group to a protein substrate. The other subunit is an activator of the catalytic subunit and is called a cyclin; the abundance of this protein increases and decreases during the cell cycle (i.e., the protein concentration cycles up and down during the cell cycle). Different cyclins are specific for various CDKs and for the different phases of the cell cycle. The various cyclins are identified by letters, such as cyclin A and cyclin B. Cyclins must reach a threshold concentration to activate the catalytic subunit, and the threshold is achieved as a result of protein accumulation from new synthesis during the G phases. The different phases of the cell cycle are controlled by different cyclin-CDK pairs, as shown in Figure 2-4. Thus the complex of CDK1 with either cyclin B or cyclin A is the particular CDK pair responsible for driving the cell into mitosis. Cyclins E and A interacting with CDK2 play important roles in initiating and maintaining DNA synthesis in S phase. Cyclin D interacting with either CDK4 or CDK6 functions in late G1 in a “decision” by the cell to commit to DNA synthesis. This decision is called the restriction (R) point and is discussed in the later section on tumor suppressors. The environmental stimulatory signals for cell division can be as simple and nonspecific as availability of nutrients, to the extent that cells only divide when they have approximately doubled in size through synthetic growth. However, two more specific stimulators of the cell cycle are primarily implicated in cancer. One is the response to soluble growth factors found in the circulation and in the extracellular fluid surrounding cells (see Chapter 1). Growth factors are proteins secreted by a variety of other cell types that are required for the division, and indeed survival, of normal, noncancerous cells. Cancer cells, however, can divide and survive with little or no stimulation from growth factors because of the acquired ability to synthesize growth factors of their own, or the activation of downstream elements in the signaling pathway. Further analysis revealed that these oncogenes often encode normal stimulators of the cell cycle, and the mutations involved had the effect of permanently activating an element in the cell cycle pathway. You can see how this would work based on the Rube Goldberg cartoon of Figure 1-13. Note that all the elements in the garage door opener are stimulatory; if any one turns “on,” a signal is sent “downstream” to cause the garage door to open. If the fish tank of the cartoon were to “mutate” by developing a leak, an “on” signal would be sent downstream of the fish tank, regardless of whether a car had pulled into the driveway. So it is with the oncogene elements controlling the cell cycle. If one of the elements mutates to turn itself “on,” that is, acquired a gain-of-function mutation, it will stimulate cell division and contribute to cancer. To return to the automobile analogy, oncogenes represent a stuck throttle or accelerator pedal. The normal, well-behaved versions of the oncogene (a watertight fish tank before the bullet, Figure 1-13) are called proto-oncogenes. Thus, strictly speaking, oncogenes have their normal equivalent as proto-oncogenes. However, given this awkward usage, increasingly the normal versions are also informally called oncogenes, and it is usually clear from the context whether the mutant or normal version is being discussed. The molecules and molecular events of the oncogene pathway (also called the growth factor or MAP kinase pathway) are discussed later. The growth factor/oncogene pathway begins with growth factors that function in a familiar way, as discussed in Chapter 1: they bind to and activate an integral membrane protein receptor. Indeed, growth factor receptors belong to the third family of receptors for environmental signals, the receptor tyrosine kinase family. This family of signal transducers has some similarities with the G-protein–coupled receptors (GPCRs), but also some important differences. Receptor tyrosine kinases (RTKs) do not require second messengers, but they do function through protein kinase activity (as many GPCRs do). The structure of RTKs is such that binding of ligand (a growth factor) by the extracellular portion of the receptor directly activates protein kinase activity by the cytoplasmic portion of the protein. The receptor itself is an enzyme (Figure 2-5). Thus the RTK carries the message across the plasma membrane, without the need for a second message. RTKs specifically add a phosphate group to a tyrosine residue of the substrate protein. This differs from the protein kinases discussed in Chapter 1 (PKA and PKC), which add the phosphate to serine or threonine residues. Phosphorylation of tyrosine residues within a protein is largely (but not exclusively) specialized to control cell growth pathways, and therefore tyrosine kinase activity generally is associated with stimulation of proliferation. The growth factors that bind to the RTKs are too diverse to be discussed at length in this chapter. Rather, one important similarity for introductory professional students is that these factors are all poorly named, so do not judge the factor by its name. Sometimes growth factors have “growth factor” in their name; some are referred to as cytokines; and some are called colony-stimulating factors (for growth of colonies in soft agar, as previously mentioned). Further confusion arises because their names always reflect their history but rarely their broader function. Thus, “epidermal growth factor” stimulates cell division in many more types of cells than only skin cells, but it was discovered using skin cells. The other, more important similarity among growth factors is that whatever their name they share a conserved basic pathway and “strategy” for control, as with the numerous ligands binding GPCRs and nuclear receptors, of their downstream effectors, in this case the CDK engines of the cell cycle. Growth factor activation of RTKs stimulates a pathway involving a G-protein “on-off” molecular switch, the Ras protein introduced in Chapter 1, and uses a cascade of protein kinases, both tyrosine and serine-threonine, called the MAP kinase pathway. Ultimately, the MAP kinase pathway activates transcription factors, in turn controlling the expression of cyclins, and other direct regulators of CDKs (see Figure 2-5). After activation of the RTK, the next major step in the growth factor/oncogene pathway in normal cells is activation of the protein product of the ras proto-oncogene. Investigations of how it worked revealed that the Ras protein was an important member of the small G-protein family of molecular regulators, all of which have intrinsic guanosinetriphosphatase (GTPase) activity and serve as molecular “on-off switches.” These proteins control many basic cellular functions, and the heterotrimeric G protein evolved from Ras-like ancestor proteins (see Chapter 1). Indeed, in yeast it is Ras, not a heterotrimeric G protein, that controls adenyl cyclase and phospholipase C (see Figure 1-18). Figure 2-6 illustrates the duty cycle of this on-off switch and its basic similarity to the alpha subunit (Gα) of the heterotrimeric G proteins. Ras, other small G proteins, and Gα all are in the “on” state when they have guanosine triphosphate (GTP) bound to them (because of receptor activation). All are in the “off” state when the G protein hydrolyzes its GTP so that guanosine diphosphate (GDP) is now bound. You can see how this gene could be discovered as an oncogene, that is, a gene in which a gain-of-function mutation contributes to the development of cancer. If the GTPase activity is lost by mutation, this simple, enzymatic on-off switch remains trapped in the “on” position (the accelerator pedal is stuck). It continues to send an activating signal to the downstream cell cycle machinery without the presence of growth factors or the activation of RTKs. In fact, such mutations in Ras underlie its oncogenic function, and it is estimated that 30% of human cancers have activating mutations in their ras gene. Other small G proteins control a myriad of cellular functions, including others involved in cancer. Thus the Rho subfamily of small G proteins is directly involved in the spread of cancer because it helps regulate actin assembly and activity. As described later, the spread of cancer depends on the ability of cells to migrate through tissues. This “crawling” motility in turn depends on a musclelike mechanism based on actin and myosin (see Figure 1-4). Although the basic, on-off activity of Ras and Rho are the same as that shown in Figure 2-6, Rho is connected to actin, whereas active Ras activates the elements of the MAP kinase pathway. GTP-bound Ras causes the sequential activation of a series of protein kinases, called Raf, Mek, and Erk. Raf phosphorylates and activates Mek, which in turn phosphorylates and activates Erk, as shown in Figure 2-5. This trio of kinases is called the mitogen-activated protein kinase, or MAP kinase, pathway (a mitogen is a stimulator of mitosis, e.g., a growth factor). If any of these three protein kinases should experience a gain-of-function mutation irreversibly activating the protein kinase, a stimulatory signal is sent down the remainder of the pathway. Thus, as with ras, these three kinase genes act as oncogenes. Raf, Mek, and Erk are a specific example of yet another conserved but diverse general module of information transduction. There are MAP kinase trios other than Raf, Mek, and Erk. Although it is not worthwhile to give names to all the various specific pathways, it should be noted that these trios have a systematic set of names for their elements. Raf is a MAP kinase, kinase, kinase (a MAPKKK). Mek is a MAP kinase, kinase (MAPKK), and Erk protein is the MAP kinase (MAPK) itself. This jargon is awkward, but it is widely used and logical, as Figure 2-5 suggests. When activated, Erk activates one or more transcription factors that control the transcription and translation of a key regulator of the cyclin-CDK engine. One of these transcription factors, Myc (“mick”), is encoded by another important oncogene/proto-oncogene. As with ras, the myc gene is mutated in a high frequency of human tumors, giving rise to an oncogenic form able to activate the cell cycle. As shown in Figure 2-5, Myc protein is involved in the transcription of a variety of cyclins and of the CDK2 catalytic subunit and plays a significant role in allowing the cell to pass from G1 to S phase. Myc is also involved in many other transcription events related to cell growth, differentiation, and cancer. Both cell-cell and cell-ECM adhesion activate the MAP kinase pathway, similar to growth factors, but the Ras intermediate is less important here. Figure 2-7 shows the activation of the MAP kinase pathway as a result of cell-ECM adhesion. The adhesion receptors that bind to ECM are called integrins and these activate the MAP kinase pathway via two important intermediates that are oncogenes. One is Src (“sark”), a protein tyrosine kinase and the first oncogene (src) to be discovered. Unlike the RTKs previously described, Src is not a receptor. However, Src is located on the inside face of the plasma membrane, where it can interact with adhesion receptors. Another important intermediate is also a protein tyrosine kinase, called Fak (focal adhesion kinase). As before, activation of Src and Fak activate the MAP kinase pathway, leading to increased cell division. Again, mutation or overexpression of src and fak sends inappropriate stimulation to the cell cycle machinery, which facilitates cancer. As mutant oncogenes, fak is associated with aggressive melanomas in humans. The src oncogene was named because of its ability to cause sarcomas in chickens.
Cancer
A Disease of Cellular Proliferation, Life Span, and Death
Cancer Arises from Genetic Dysfunction in the Regulation of the Cell Cycle, Cell Life Span, and Cell Suicide
Control of the Cell Cycle (Proliferation)
Cell Division Is the Result of a Clocklike Cell Cycle
Cyclin-Dependent Kinases Are the “Engines” Driving the Cell Cycle
The CDK “Engines” Are Controlled by Both Throttle (Oncogene) and Brake (Tumor Suppressor) Controls
Growth Factor Pathway: Stimulator of Cell Proliferation
The Cell Cycle Is Stimulated by Growth Factors that Bind to and Activate Receptor Tyrosine Kinases
The Ras Oncogene Contributes to Many Cancers and Serves as a Model for Understanding Small G Proteins
The MAP Kinase Pathway Leads to the Expression of Cyclins and Other Stimulators of the Cell Cycle
The MAP Kinase Pathway also Mediates the Stimulation of the Cell Cycle by Cell Adhesion
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Cancer: A Disease of Cellular Proliferation, Life Span, and Death
Only gold members can continue reading. Log In or Register to continue
