Chapter 8 Ascites
Definition
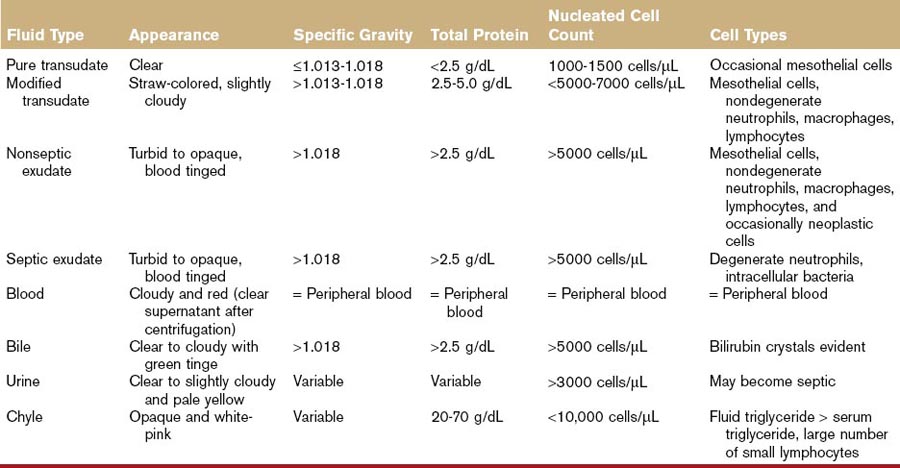
Ascites is defined as fluid accumulation within the peritoneal cavity. Although any peritoneal fluid accumulation could be classified as ascites, the term is more often used to refer to pure and modified transudates. Pure transudates are clear, colorless fluids, with low nucleated cell count (< 1.0 to 1.5 × 109 cells/L or <1000 to 1500 cells/µL), specific gravity ≤1.013 to 1.018, and total protein <2.5 g/dL (<25 g/L). Modified transudates may be clear, straw colored, or cloudy in appearance. The protein count of a modified transudate is slightly higher (2.5 to 5.0 g/dL or 25 to 50 g/L) as is the total nucleated cell count (<5.0 to 7.0 × 109 cells /L or 5000 to 7000 cells/µL) and specific gravity (>1.013 to 1.018).1 Exudates are viscous and/or serosanguineous in appearance, have a high protein content (>5.0 g/dL) and cellularity (>10,000 cells/µL), and have a relatively high specific gravity (>1.018). Ascites generally indicates serious disease and warrants prompt investigation.
Pathophysiology and Mechanisms
Ascites forms because of disruption of Starling forces in local capillary beds, with diffusion of fluid across the serosa into the peritoneal cavity. Under normal circumstances, portal pressure gradient decreases (from 6.5 to 13 cm H2O to 3 to 3.5 cm H2O) as it progresses from the hepatic triads through to the sinusoids, central vein, hepatic veins, and the caudal vena cava.2 The changing pressure gradient pushes fluid from sinusoids into protein-rich perisinusoidal spaces (spaces of Disse) which are covered by a thin membrane.3,4 The balance between oncotic and hydrostatic pressures is maintained by lymphatic system (much of it located in the diaphragm) returning tissue fluid to the vascular system.5,6
Portal hypertension is a sustained increase in blood pressure of portal vasculature. Portal hypertension can be caused by capillarization of hepatic sinusoidal endothelium, increased portal blood volume, or increased resistance to normal flow.3,6,7 Diseases that lead to portal hypertension are divided anatomically into prehepatic, intrahepatic (presinusoidal, sinusoidal, or postsinusoidal), and posthepatic causes.7–9 Ascites in posthepatic and sinusoidal or postsinusoidal intrahepatic portal hypertension develops when sinusoidal pressure increases so that fluid leaving sinusoids exceeds the capacity of lymphatics to return it to circulation.8 The fluid that leaves the surface of the liver has percolated through the spaces of Disse where hepatic lymph has a high protein content, resulting in formation of a modified transudate.6,10,11 Acquired portosystemic shunts do not develop in posthepatic portal hypertension because caval pressure also increases significantly.7,8
Ascites caused by presinusoidal intrahepatic and prehepatic portal hypertension is uncommon because the rate of fluid production rarely exceeds rate of lymphatic absorption.6,7,9 When accumulation exceeds absorption, a pure transudate develops because there is transudation of intestinal lymph which has a much lower protein content than hepatic lymph.9,12
Regardless of cause or anatomic site of portal hypertension, the renin–angiotensin–aldosterone system (RAAS) contributes to ascites formation.13 Circulating blood volume may be initially increased by development of extrahepatic portosystemic shunts and shunting of blood to systemic circulation.14–16 When portal hypertension exceeds this capacity, there may be a decrease in circulating intravascular blood volume detected by volume receptors,3,8,13,17 and the RAAS becomes activated. Decreased renal prostaglandins and natriuretic factor result in water and sodium retention.6,7,18 Further increases in blood volume perpetuates the tendency toward ascites formation.19–22 If portal pressures are already elevated, altered renal sodium handling may be a precipitating (rather than a perpetuating) factor for ascites formation.23–25
Increases in capillary hydrostatic pressure resulting from intravenous fluid overload or reduced cardiac output may occasionally be severe enough to induce ascites, although pulmonary edema is often the predominant clinical sign particularly in cats.1 Vasculitis is another potential cause of fluid accumulation in abdominal and thoracic body cavities.
Differential Diagnosis
Posthepatic portal hypertension occurs when there is obstruction of flow from hepatic veins and caudal vena cava to the right side of the heart. Pericardial disease (e.g., pericardial effusion or pericarditis causing cardiac tamponade) is the most common cause in dogs.26–28 Other possible causes include right-sided cardiac disease (e.g., intracardiac masses, tricuspid valve insufficiency, cor triatrium dexter, pulmonic stenosis) and pulmonary hypertension (e.g., primary or caused by chronic respiratory disease, left-sided congestive heart failure, pulmonary thromboembolism and dirofilariasis).29–33 Obstruction of the caudal vena cava because of congenital abnormalities, thrombi formation, dirofilariasis (“caval syndrome”), neoplasia, or trauma may also occur.34–37 Budd-Chiari–like syndrome refers to obstruction of hepatic venous flow to the right atrium.38 Posthepatic portal hypertension is uncommon in cats.1
Hepatic (sinusoidal or postsinusoidal) portal hypertension generally results from chronic hepatic disease that either increases resistance to portal blood flow or increases hepatic arterial blood flow. The most common cause of sinusoidal or postsinusoidal portal hypertension is diffuse parenchymal disease such as cirrhosis (an end-stage disease in breed-specific hepatopathies), drug-related hepatopathies, fibrosis, diffuse neoplasia, chronic diffuse hepatitis, and chronic cholangiohepatitis.6,15,39,40 In rare cases, severe hepatocyte swelling due to vacuolar hepatopathy (e.g., hepatic lipidosis) may cause portal hypertension, but rarely does it cause fluid accumulation.1
Prehepatic or presinusoidal hepatic portal hypertension results from intraluminal occlusion of the portal vein (thrombosis, neoplasia), abnormal developmental anomalies of the portal vein (stenosis, atresia, hypoplasia) or extraluminal compression of the portal vein by neoplasia or other abdominal masses.41–44 It may also develop as a consequence of surgical ligation of a congenital portosystemic shunt.1 Congenital arterioportal fistula is a direct communication between a portal venous branch and a hepatic arterial branch, representing another presinusoidal portal hypertension.45,46
Hypoalbuminemia is another cause of ascites formation, when serum albumin concentration falls below 1.5 g/L. Albumin may be decreased because of excessive losses, decreased production, or protein-calorie malnutrition. Albumin is a small protein (molecular weight 66,000 daltons) easily lost in severe glomerular disease (protein-losing nephropathy), as well as protein-losing enteropathy and severe exudative cutaneous lesions. Albumin may decrease in other exudative process such as septic or neoplastic peritonitis. The liver is the major site of albumin synthesis and is capable of producing adequate albumin at 33% of maximal capacity.47 That factor, in addition to the relatively long circulating half-life of albumin (8 to 9 days) means that hepatic disease must be both chronic and severe to cause clinically important hypoalbuminemia.6,47,48 End-stage acquired hepatic disease, and congenital portosystemic shunting both cause substantially decreased albumin production.42 Decreased nutritional intake alone is seldom sufficient to cause albumin to decrease to such an important degree, but starvation will contribute to the severity of hypoalbuminemia associated with other causes.
Evaluation of the Patient
History
History is highly dependent upon underlying disease. Congenital diseases would be more likely in young animals, while some breeds are predisposed to specific hepatopathies, congenital portosystemic shunts, portovascular anomalies, or hepatic fibrosis.46,49,50 Owners may present their animal for “weight gain” caused by abdominal distention. If ascites is caused by primary hepatic disease, the animal may display neurobehavioral signs ranging from changes in temperament to seizures and coma caused by hepatic encephalopathy. Other signs of hepatic dysfunction may be present such as anesthetic or sedative intolerance, dermatoses, polyuria/polydipsia, melena, acholic feces, vomiting, diarrhea, weakness, and lethargy.47,51 Determination of toxin and drug exposure is important.
Dyspnea, exercise intolerance, and syncope may be present if concurrent pleural effusion is present. Dyspnea could still be a manifestation of ascites if abdominal fluid volume impedes diaphragmatic muscle contractility.52 Heartworm prophylaxis status should be established in animals from heartworm endemic areas. Losses of albumin through exudative processes are fairly self-evident, but clinical signs associated with loss through glomeruli or intestines may be less obvious. Small bowel diarrhea may be present or absent even in severe small-intestine disease if the large intestine retains sufficient capacity to absorb excess fecal water. Establishment of intestinal parasitic prophylaxis may also be important, especially in young puppies or kittens.
Physical Examination
Expected physical examination findings are also dependent on underlying disease processes. In general, ascites can be detected only when a large volume of fluid is present. This large volume often precludes thorough abdominal palpation and assessment of organ size. Animals with profound abdominal distention may have loss of dorsal musculature and body condition. Assessment of mucous membrane color is often useful and may demonstrate anemia (Chapter 16) or icterus (Chapter 18). Prolonged capillary refill time or skin tenting may be present with dehydration, and rarely there may be signs of a bleeding diathesis because of coagulopathy (Chapter 9). Digital rectal examination may provide evidence of melena, acholic feces, or hematochezia. Abdominal pain may be present with gastrointestinal ulceration or perforation.
Laboratory Evaluation and Tests
Figure 8-1 outlines an algorithmic approach to diagnostic evaluation of ascites. The first step in diagnostic evaluation is abdominal fluid analysis to allow differentiation of pure and modified transudates, septic and nonseptic exudates, blood, and chyle. Abdominocentesis is readily performed in conscious animals and should be performed in a sterile manner with needle insertion at the level of the umbilicus, just lateral to midline. Peritoneal lavage is rarely required in cases of ascites. If there is a suspicion of concurrent pleural effusion, then thoracocentesis may be performed in preference to abdominocentesis as both a palliative and diagnostic tool.
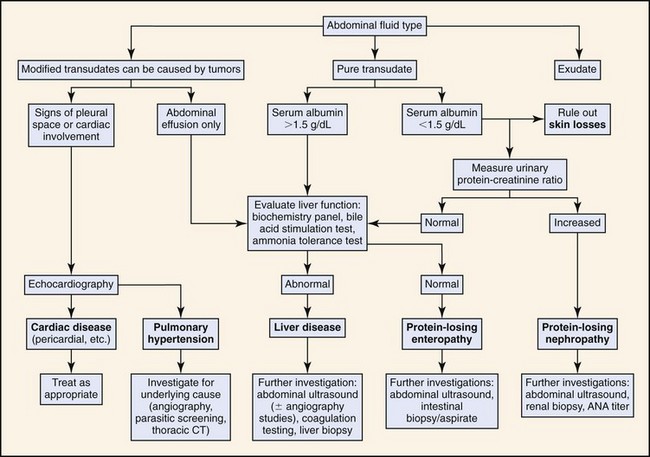
Ascitic fluid is first characterized according to appearance, specific gravity, total protein, and cellularity. Table 8-1 outlines the basic differences between fluid types. Fluid analysis should include cytologic and chemical evaluation. Additional fluid analysis could include Gram staining and culture (bacterial infection), measurement of blood urea nitrogen (BUN) and creatinine (renal failure), and triglyceride analysis (lymphatic transport disorders). Determination of serum-ascites albumin gradient may be used to differentiate between transudative and exudative processes. A serum-ascites albumin gradient >1.1 g/dL is usually indicative of portal hypertension but may not distinguish between hepatic and posthepatic portal hypertension.53 Ascitic fluid amino acid profiles (e.g., glutamate-to-glutamine ratio) may help to differentiate cirrhosis and other noncirrhotic causes of ascites.54
< div class='tao-gold-member'>
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


