(1)
Neurosurgical Foundation in Providence, Providence, RI, USA
Abstract
Normal Pressure Hydrocephalus (NPH) is a syndrome of dementia, gait disturbances, and urinary incontinence affecting the elderly population. Neuroimaging shows ventriculomegaly, and from the 70s to the early 90s invasive testing of intracranial pressure (ICP) and cerebrospinal fluid (CSF) dynamics supported the concept of a disturbed CSF circulation in those patients. Implantation of ventricular shunts has shown considerable symptomatic improvement in individuals; however, age-related comorbidity and coexisting dementias, such Alzheimer’s disease (AD) and subcortical arteriosclerotic encephalopathy (SAE), have conflicted both clinical diagnosis of the syndrome and outcome to treatment. Moreover, a pathological continuum co-existing in the dementias of SAE, AD, and NPH has been proposed more recently, as blood flow and CSF biomarker studies as well as brain biopsies taken at the time of shunt implantation have evidenced the role of chronic ischemia and white-matter disease in NPH patients and biopsies were classified as “Probable AD” according to the Consortium to establish a registry of Alzheimer’s disease (CERAD). Aging has shown to be a main risk factor to NPH, but also to AD and SAE, and as the population is aging in both the developed and developing countries, the prevalence of NPH dementia is very likely to increase. As such, research aspects aim at an understanding of the comorbidity, and how it impacts the diagnosis, pathophysiology, and the outcome of NPH. Certainly, NPH has to be seen from the multi- and transdisciplinary perspective, and experimental models are needed to model the interaction of hydrocephalus with risk factors of aging, and the coexisting dementias. The kaolin-induced hydrocephalus model allows preselection of age, manipulation of the disease progress in animals, and modeling of a chronic and long-term hydrocephalus condition, as existing in NPH. Kaolin, an inert silica derivate, creates a CSF malabsorption through scar formation, when injected into the subarachnoidal cisterns. A progressive ventricular enlargement follows a transient increase in ICP, ventricles remain enlarged while ICP normalized, similar to the human condition. Kaolin hydrocephalus induced in the aging 12-months-old rat has supported both, AD- and SAE-pathological correlates the longer the hydrocephalus existed in the animals: a mild but chronic “sublethal” ischemia has been evidenced by C-14-Jodo-Autoradiographic blood flow studies, and intracerebral and perivascular accumulation of Aβ 1–40, Aβ 1–42 peptides and hyperphosphorylated TAU proteins were observed through qualitative and quantitative histology. An age-related CSF circulatory dysfunction and reduced CSF turnover with a subsequent failure to clear Aβ 1–40, Aβ 1–42, and other toxic metabolites out from the brain interstitial fluid was therefore suggested as a common pathological element in NPH and NPH coexisting with AD and SAE. Further evidence to a common element of metabolic failure existing in those dementias was supported by significant breakdown of blood-brain-barrier (BBB) receptors in the aging Kaolin-rat, the best characterized being the low-density-lipoprotein-related protein 1 (LRP-1), which allows Aβ to escape the brain through the capillary border. With aging, Aβ LRP-1 transport is reduced by 50%.
Finally, shunt treatment in Kaolin-hydrocephalus has shown that “irreversibility” in the hydrocephalic brain lies in the damage of “ischemia vulnerable” brain regions, such as the CA1 sector of the hippocampus. Changes in synaptophysin and neurofilament immunohistochemistry of CA1 have correlated with histological changes of “delayed neuronal death” and occurred early in the course of hydrocephalus. This warrants a timely shunting of patients. The studies in aging kaolin-induced hydrocephalus might allow insight into late-life disturbances in CSF circulation, CSF turnover, and BBB transport underlying several age-related dementias, including AD. This reconsiders the traditional concept of “shunts for dementia”; however, both clinical and experimental hydrocephalus research should embrace the multidisciplinary aspect as it might allow and bring further insight into the interaction of ICP, impaired CSF circulation, blood flow, and metabolic breakdown in the various dementias.
Key words
Normal pressure hydrocephalusKaolin-hydrocephalusintracranial pressureCSF circulationshunt treatment1 Introduction
1.1 Dementia in Normal Pressure Hydrocephalus
Normal pressure hydrocephalus (NPH) was first described by Hakim and Adams in the 1960s (1) as a syndrome consisting of dementia, gait disturbance, and urinary incontinence. Patients exhibited enlarged ventricles; however, there was absence of raised intracranial pressure (ICP). The initial series, which reported three cases, two posttraumatic and one idiopathic without known cause, encouraged publications in the 1970s and early 1980s on “Ventricular shunts for dementia” (2, 3) as Hakim went on to describe the “classic triad” of gait disturbance, incontinence, and dementia, which were improved with the removal of cerebrospinal fluid (CSF) (4, 5). However, in the 1980s and 1990s, the results on larger patient series treated accordingly were frustrating as the outcome was affected by variable improvement after shunt treatment. Despite considerable high percentages (20–30%) of surgical risks and persistent morbidity (6), long-term results were poor (7). Particularly in idiopathic NPH, long-term prognosis was affected by many comorbidities not related to the shunt procedure, such as cerebrovascular and cardiovascular disease and other age-related illnesses (7–13).
Although no definite epidemiological data exist (14), we are presently facing a situation of patient numbers increasing for both idiopathic NPH and secondary NPH patients in many neurological and neurosurgical services, in part, as a result from the improved healthcare and increased longevity. Calculations based on focused centers, such as memory or dementia clinics, estimated both incidence of NPH among the elderly population and prevalence of NPH in dementia ranging from 4% to 14% (15, 16). From own experiences, patients in their 80s are now seeking an improved quality of life, and despite the risks of surgery, are no longer willing to accept the age-related burden. As such, efforts are being made towards a more accurate diagnosis and a more individual “differential” treatment of those patients (17).
However, one of the main obstacles is that the pathophysiology of NPH is still poorly understood. The traditional concepts of a “CSF malabsorption” and the “Intracranial Pressure–Volume Relationship” (18–22) have failed to explain the whole spectrum of the disease, e.g., the origin and prognosis of the gait and cognitive symptoms, as well as the significance of the vascular pathology and cerebral atrophy as seen in imaging studies and autopsies (23–27) – Fig. 1. Also, measures of resistance to CSF outflow (Rout) and/or ICP-volume index, analysis of ICP oscillations (“B-waves”) have shown invalid in the diagnosis of NPH (28), although those measures have shown an association with chronic hydrocephalus and CSF malabsorption in earlier studies (29). Moreover, neuropathological findings, such as arachnoid fibrosis or vascular pathology, obtained from biopsies taken at the time of shunt surgery in patients diagnosed with NPH, did not show any correlation of an increased Rout or disturbed pressure profile as evidenced by B-waves (30, 31).
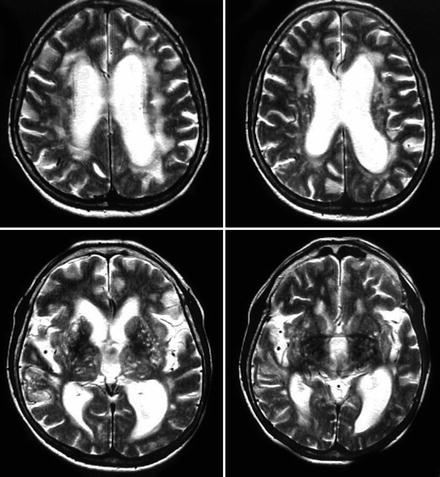
Fig. 1.
Sagittal T2 MRI of idiopathic NPH. On axial T2-weighted magnetic resonance images deep white-matter and periventricular white-matter hyperintensities/lesions are frequently seen in idiopathic normal pressure hydrocephalus indicating cerebrovascular comorbidity. Around 80% of idiopathic NPH patients have vascular risk factors [8]. These lesions are not exclusion criteria for the diagnosis of NPH and decision-making for shunt treatment.
Neuroimaging, blood flow, brain metabolic studies, and analysis of CSF biomarkers have enlightened the presence and role of ischemia and metabolic failure in the NPH syndrome (32–41). Impairment of cerebrovascular reserve capacity (26 27 42), blood flow reductions, and reduced metabolism in frontal, supplementary motor, and anterior cingulated areas, as well as temporomesial, limbic areas have correlated with both the motor and cognitive dysfunction before and with changes after shunt treatment (32, 39, 43). Other PET-studies, using an anatomic region-of-interest analysis on coregistered MRI done in both secondary and idiopathic NPH, demonstrated cerebral blood flow reduction in the basal ganglia and the thalamus. This was suggested to correlate with the level of motor dysfunction in patients (33). Specific disturbances in regional brain metabolism have also been evidenced by 1H- and 31P-MRI Spectroscopy investigations in NPH patients (38): lactate peaks in periventricular areas and reduced cortical N-acetyl-aspartate/total creatine (NAA/tCr) ratios indicating neuronal disintegration and mitochondrial dysfunction have been shown and differed from controls and other forms of dementia. Microdialysis studies before and after high-volume CSF withdrawal have indicated an improvement in tissue oxygenation and increased glucose levels while glutamate levels decreased, comparable to a situation of postischemic recovery in patients (44).
Those measures have allowed access to the brain disease and the dementia in hydrocephalus, which is not simply a matter of ventriculomegaly, ICP, and “disturbed CSF-dynamics.”
1.2 Comorbidity of Dementia in Idiopathic NPH
Of importance for an improved diagnosis and treatment of NPH is an understanding of the comorbidity, and how it impacts the pathophysiology and the outcome of the disease. Among the most important coexisting diseases are Binswanger’s (Subcortical arteriosclerotic encephalopathy, SAE) and Alzheimer’s dementia (AD). They exhibit clinical and imaging features in common with NPH (8, 23, 31, 45) and the above-mentioned series on neuropathological changes consistent with AD on cortical biopsies have further suggested that the two diseases may have some common pathophysiology (46, 47). Seven of 21 NPH patients (33%) in one series had neuropathological changes consistent with AD on cortical biopsy at the time of shunt placement (48). In a second series, one-third to one-half of NPH patients had Alzheimer-type neuropathology in cortical samples obtained at shunt implantation with dramatic clinical improvement (49) and, in a more recent series, 23 of 55 patients biopsies at the time of shunt placement for NPH were classified as “Probable AD” according to the CERAD (Consortium to establish a registry for Alzheimer’s disease) criteria, including 75% of those with severe dementia (50). This coincidence of Alzheimer-type neuropathology among patients with NPH is greater than one would anticipate if the two diseases were unrelated. It was suggested, therefore, that the two diseases have a common basis in an age-related CSF circulatory dysfunction and a reduced CSF turnover with a subsequent failure to clear the various substances resulting from glial and neuronal metabolism out of the brain interstitial fluid (46, 51). These observations are supported by recent studies on elevated CSF contents of neuronal degrading products (hyperphosphorylated Tau (hpTau), amyloid-β (Aβ), and neurofilament) as well as biomarkers reflecting a disturbed neuronal metabolism, e.g., neuropeptides and transmitter-degrading products (41).
Among the neuronal CSF biomarkers, neurofilament protein has shown to be increased in idiopathic NPH, when compared to other dementias (52) or age-matched controls (53). Moreover, neurofilament levels correlated with the degree of dementia and cognitive impairment (“impaired wakefulness”), as well as improvement of cognition after shunt treatment (41, 54). Furthermore, reduction of neurofilament protein levels in both the lumbar and ventricular CSF after shunt treatment have correlated with a reduction of “irregular-type” periventricular white-matter hyperintensities seen in frontal T2-weighted turbo spin-echo sequences in patients with improving symptomatology (55). While neurofilament seems a marker of “reversible” damage in the human condition, sulfatide contents in NPH correlated with the degree of ischemia and “irreversible damage” in patients, as they were associated with poorer outcome to shunt treatment (40). However, there was also a clear overlap in CSF sulfatide measures comparing 43 patients with NPH and 19 patients with SAE, supporting the pathological continuum between both dementias (40).
2 Animal Models of Hydrocephalus and NPH
Different from human hydrocephalus and NPH, changes in the neuronal and glial metabolism have been largely described in animal models of hydrocephalus (56). Blood flow reductions in subcortical and cortical areas (57, 58), as well as mild but prolonged and chronic ischemia (59) have been evidenced. Here, the experimental results have much more straightforwardly evidenced that in the chronic hydrocephalic state cerebrovascular adaptive processes, e.g., angiogenesis and neuronal plasticity, might play a more important role in the disease process than ventricular enlargement and CSF dynamics (59, 60). Changes in various metabolic markers as well as cholinergic and dopaminergic transmitters have been also correlated with behavior and learning ability in chronic adult hydrocephalus (61–63).
In general, hydrocephalic models fall into the categories of obstructive, teratogenic, infectious, and genetically modified. Teratogenetic (e.g., vitamin-deficient, cytostatics), viral-induced, and genetic models, which are naturally occurring or induced, are most frequently prenatal or neonatal in onset (64–66). The most frequently used genetic models are the H-TX (67–69) and the LEW/Jms rat, both prenatal multigenic (70–72), and the Hy-3 mouse (73), where gene abnormality is present in hydin, a gene controlling cilia and flagella. However, all the infectious, genetic, and teratogenic models do not provide adequate models in the study of the significance of neurodegeneration and treatment options in the human chronic hydrocephalus condition (70). As to their pre- or neonatal onset and the developmentally acquired and toxic nature, as well as to the rapid development of ventriculomegaly, hydrocephalus, and ICP increases and detrimental effects, they do not apply to the generally mild and chronic onset of symptoms and disease duration and the mild to moderate ventriculomegaly, e.g., as observed in NPH, and also in other types of adult-or late-life-onset hydrocephalus (74, 75). Also, the coexisting complex brain malformations and systemic diseases and failures conflict the use of those models.
Even though some recent observations suggest a genetic linkage in a small subpopulation of patients with NPH (76, 77), the existing genetic-induced models are not considered to be of significance for the investigation of NPH-related dementia.
The most common method for creating hydrocephalus allowing a preselection of age of onset, the severity of ventriculomegaly and disease course are models of mechanical or chemical obstruction of the CSF pathways. Obstruction is created at the level of the aqueduct, the fourth ventricular outlets, the basal cisterns and/or venous system by injection of cyanoacrylate-gel (78, 79), Kaolin (80, 81), an inert silica derivate, or Silastic Oil (82–86). Ventricular dilatation is variable depending on the material and site of injection and obstruction. Furthermore, dilatation and its effects are more severe in younger animals (87, 88).
Those animal models mimic various forms of “obstructive” hydrocephalus; however, in light of the human condition, in idiopathic and secondary NPH, mostly a communicating type of hydrocephalus characterizes the chronic hydrocephalus disease (e.g., after trauma, hemorrhage, or meningitis), at least according to the classical definition of “communicating” versus “noncommunicating” hydrocephalus (89, 90).
Several attempts have been made in the past 20 years to induce communicating hydrocephalus in dogs, rats, and monkey by injecting kaolin, silicone, or Silastic oil into the subarachnoidal space (SAS) or the overlying cerebral hemispheres. However, the applied methods do not produce the disorder consistently and the site of obstruction has not been determined unequivocally. Nonmechanical induction methods and models, such as blood injection, bacterial inoculations, intrathecal injections of growth factors (e.g., transforming growth factor beta -TGF-β), and neurotoxins have also been used to produce “posthemorrhagic” and “postmeningitic” communicating hydrocephalus (91). More recently, renewed attempts to produce communicating-type hydrocephalus with kaolin injections into the cortical subarachnoid space or in the prepontine cisterns of adult rats have been reported, and the affected animals have been shown to survive for several months (91, 92).
However, to date there are no adequate “natural” NPH models available. All the above-mentioned procedures include the additional influence of the inducing chemical substances or implants on the pathophysiology, and thus are not true representations of the effects of the CSF obstruction or an absorptive defect alone on the brain.
Particularly when arguing for NPH, there is a lack of models to study across the age spectrum from brain development to senescent. Models using senescent animals might improve our understanding of the interaction of hydrocephalus with risk factors of aging, and the comorbidity of hypertensive vascular disease and AD, which are common in elderly patients with idiopathic NPH (Consensus from a NIH funded meeting held in Bethesda US, September 29–30, 2005: “Hydrocephalus: Myths, New Facts, and Clear Directions”, Lit.) (93).
2.1 The Kaolin-Induced Model of the Adult and the Aging Rat
Hydrocephalus in the Kaolin models is generally induced by intracisternal injection of Kaolin suspension (aluminum silicate diluted with 0.9% saline; 1:4) under general anesthesia. In earlier studies of 3-month-old rats, 0.1 ml kaolin suspension was slowly injected through a 20-gauge needle inserted into the cisterna magna after microsurgical exposure of the atlanto-occipital membrane (59, 94, 95) – Fig. 2. The Kaolin-induced blockage of CSF circulation in the basal SAS results in hydrocephalus, increased resistance to CSF absorption, and consequent rises in ICP over a short-term period of 2 weeks. However, over a long-term period above 4–6 weeks, ICP normalizes, while resistance to CSF outflow remains above normal values (94). This is paralleled by a gradual progress of ventricular dilatation from short-term to long-term, and ventricles remain enlarged despite normalization of ICP, quite similar to the dynamics observed in NPH and adult chronic hydrocephalus (29)– Fig. 2.
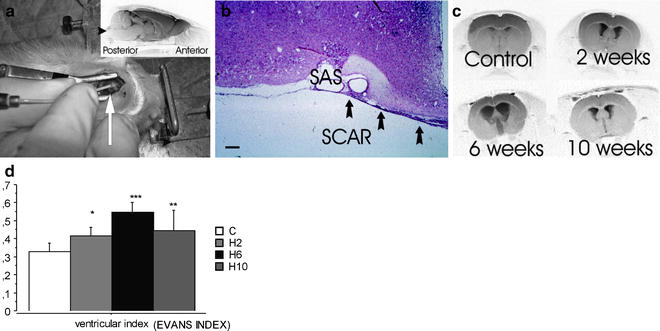
Fig. 2.
Kaolin-induced hydrocephalus in the adult (3-month-old) rat. (a) Photograph of a Sprague–Dawley rat with the head mounted in a stereotaxic frame. Arrow points to catheter inserted into cisterna magna right below the exposed occipital bone. Inset shows lateral MRI view of rat brain (4.7 T BRUKER Biospect). Arrowhead points to cisternal site of kaolin injection. (b) H&E photomicrograph (bar = 500 µm) showing the scar formation at the site of kaolin injection in basal subarachnoidal space (SAS). The aluminum in kaolin is bound to silicate, and therefore not free to diffuse widely. As indicated in the figure, the kaolin suspension remains confined to basal SAS. Consequently, the aluminum does not distribute into parenchyma. (c) MRI (4.7 T BRUKER Biospect; RARE sequence) coronal section showing progressive ventricular dilatation over the course of hydrocephalus. Normal pressure hydrocephalus-like changes are consistently observed: moderate ventriculomegaly, reduced CSF formation rate, and increased resistance to CSF outflow. The induced hydrocephalus is well advanced at 6 weeks postinjection. This Sprague–Dawley model therefore mimics human NPH. (d) Evan’s Index (as defined by the quotient of the largest ventricular diameter divided by the width of the brain at the level of the anterior commissure) measured in the MRI scans of the hydrocephalic rats at the assigned time points (n = 16 controls; n = 5 at 2 weeks, n = 6 at 6 weeks, n = 7 at 10 weeks; mean ± SD,* p < 0.05, ** p < 0.01, *** p < 0.001).
Kaolin induces an unspecific inflammatory response in the basal cisterns. However, beyond the short-term period of 2 weeks, the inflammatory response was no longer observed, and scar formation dominated the basal SASs as shown by our group – Fig. 3. The findings on the regional and temporal progression and the onset of major histological changes in glial and neuronal markers in both cortical and subcortical regions, e.g. in the hippocampus (95), do no suggest a direct pathological effect of kaolin injection, as also described by others (91).
Twelve-month-old rats were used to adopt to the aging NPH population, since they reflect human early senescence above the age of 50. In the aging rats, 0.03 ml kaolin suspension was sufficient to induce hydrocephalus as evidenced by measures of the Evan’s index in the coronal histological specimen.
In both the adult and the aging rat model, studies on the vascular and AD-related comorbidity, as evidenced by the above described clinical observations, were performed by histological and immunohistological studies.
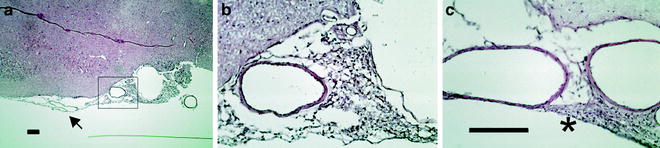
Fig. 3.
Kaolin inflammatory reaction within the basal cisterns. Photomicrograph showing kaolin-related inflammation at 2 weeks (a, b) and at 6 weeks (c) hydrocephalus in the rat subarachnoidal space (SAS). The kaolin-related inflammatory reaction is confined to the medial parts of the SAS and does not extend significantly into the more lateral cortex, as indicated by the arrow (a, bar = 500 µm). The inset shows the acute inflammatory response at higher magnification, with lymphocytes, granulocytes, and macrophages within the arachnoid granulations (b). At 6 weeks, a chronic fibrosis is seen, again in a circumscribed region of the medial basal cisterns, as indicated by the asterisk (c, bar = 500 µm). The images also display the major arteries within the SAS, which appear morphologically intact.
2.2 Histopathological Evidence of Vascular and Alzheimer-Related Comorbidity in Kaolin-Induced Hydrocephalus
Autoradiographic (C-14-Jodo-Antipyrin) studies in 3-month-old adult hydrocephalic rats have shown a prolonged decrease of cerebral blood flow in both cortical and subcortical regions ranging between 25% and 50% (59). This reflected a mild to moderate ischemia as seen in patients with cerebrovascular disease – Fig. 4. Those prolonged “sublethal, nonischemic” blood flow reductions are relevant to “silent stroke,” the pathophysiological correlate of SAE, and have shown to induce a delayed selective neuronal injury in a rat model of four vessel occlusion (96). The relevance of those prolonged “sublethal” blood flow reductions for the outcome of hydrocephalus are supported by findings of delayed neuronal injury and degeneration in ischemia-vulnerable areas in the kaolin-induced hydrocephalus model. Decreases in neurofilament and synaptophysin protein staining reactivity in the cortical pyramidal layer and the hippocampal CA-1 region have been observed, indicating an intrinsic susceptibility to cerebral hypo-per-fu-sion (59).
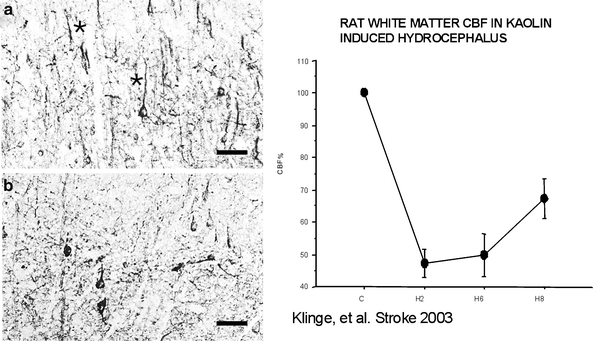
Fig. 4.
Blood-flow autoradiographic studies in kaolin-induced hydrocephalus. (a, b) Neurofilament staining in the cortical pyramidal layer IV using specific antibody against the 68 kd neurofilament subunit. In controls (a), neurofilament 68 kD was predominately located in the dendrites and also in the perikaryon of the pyramidal-type cells; * dendrite staining. In the chronic hydrocephalus stage (b), at 8 weeks, the cytoarchitecture of the pyramidal layer was disturbed. There was a loss of the regular dendrite expression of neurofilament and some of the perikarya in the superficial cortical pyramidal layer displayed a strong increase in neurofilament-expression shifted towards the cell body; bar = 25 µm (c) Examples of [14C]iodoantipyrine autoradiography brain sections (kryosections 20 µm) for control and hydrocephalic animals, in which the optical densitometric measurement of C14-acitivity was done using C14 standards. Note the weaker density in the 2 and 6 weeks examples (59). The line graph displays [14C]iodoantipyrine autoradiography cerebral blood flow (CBF) in adult rats with kaolin hydrocephalus. CBF drops by 50% in 2 weeks after hydrocephalus induction and then recovers somewhat by 8 weeks, though never returning to normal despite normalization of intracranial pressure (ICP) (error bars = 1 SE).
In light of the human findings, the neurofilament protein staining in kaolin-induced hydrocephalus also supported the observations made in patients with NPH, namely that elevated neurofilament protein in the CSF suggest a “reversible” disease state (41, 53). Transient “reactive” increase in cortical neurofilament staining in the intermediate-term were seen in periventricular cortical areas (59) before chronic “breakdown” of neurofilament staining suggested neuronal damage in the long-term stages of hydrocephalus following the chronic ischemia – Fig. 4.
Aging is the most important risk factor for developing nonfamilial “sporadic” AD (46, 97, 98) and Aβ1–40 and Aβ1–42 peptide are the most important isoforms involved in AD plaque formation and in the AD-related pathological cascade, such as hpTau formation, oxidative stress, and microglial inflammatory responses (97). In the 12-month-old rats, both peptides have been immunohistochemically observed, both in the healthy control animals and in the hydrocephalic groups; however, not in the 3-month-old hydrocephalic and control rats. This supports the role of aging as a factor contributing to Aβ pathology, both in rodents and humans (99). Also, in the Brown–Norway Fisher aging rat model (100), Aβ1–40 and Aβ1–42 accumulations have been observed to dramatically increase during senescence at an age of 36 months, which equalled the three- to fourfold “load” in Aβ1–42 in the cortex of the 12-month-old hydrocephalic rats, when counting the cortical Aβ1–42-positively stained granules (101). Aβ1–40, the more soluble of the two peptides, is usually present in the plaque periphery and stained in a diffuse, nonparticulate pattern, whereas Aβ1–42-staining was characterized by small-, medium-, and large-size Aβ1–42 granular accumulations, most likely reflecting the progressive self-aggregation of this less-soluble peptide, usually found in the plaque nidus (99) – Fig. 5. Aβ1–42 accumulations were found in and near neurons; however, most strikingly at the microvessel border in both the cortex and the hippocampus the longer the hydrocephalus persisted. Those findings translate into the cerebral amyloidangiopathy seen in dementias. It was suggested that the amyloidangiopathy plays a role in white-matter pathology, chronic ischemia, and hypoperfusion as seen in patients with vascular dementia, and vascular dementia coexisting in AD and NPH (23, 102–104).
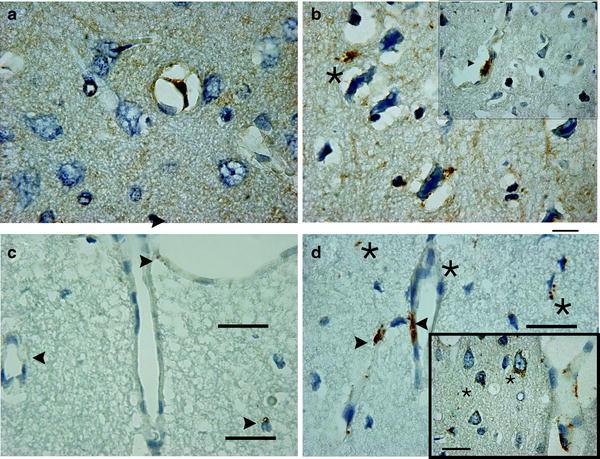
Fig. 5.
Aβ1–40 and Aβ1–42 staining in the kaolin-induced hydrocephalus model of the aging rat. Photomicrograph showing cortical Aβ1–40-positive staining in a 12-months nonhydrocephalic rat (a) and in a hydrocephalic rat 6 weeks after induction of hydrocephalus (b). In the control animals, a small amount of Aβ-positive staining is seen at a vessel border (arrowhead), whereas it is negative in the rest of the brain tissue. After 6 weeks of hydrocephalus, however, numerous Aβ1–40-positively stained granules are seen both in the parenchyma and near neurons (asterisks). Larger and more numerous Aβ1–40-positively stained deposits are also found at vessel borders (inset); bar= 25µm. Cortical Aβ1–42-positive staining in a 12-month-old nonhydrocephalic rat (c, arrowheads) and in a hydrocephalic rat 6 weeks after kaolin induction (d) shows a small amount of age-related Aβ-positive staining around cortical vessels. After 6 weeks of hydrocephalus, amyloid positive granular staining is seen at vessels (arrowheads), in and near neurons (inset) and in the parenchyma (asterisks); bar = 25 µm.
Much has been learned from the overexpression of Aβ as a result from the amyloid precursor protein (APP) cleavage induced by β- and γ-secretases, the so-called “amyloidogenic” pathway (105). On the other hand, there is Aβ metabolism characterized by a physiological clearance of Aβ peptides from the brain interstitial fluid via two main routes; (1) Clearance by diffusion and bulk flow from interstitial fluid to CSF, draining either into the lymphatic system or through the arachnoid granulations into the venous circulation (103), and (2) Aβ crossing the capillary endothelium of the blood-brain barrier (BBB) via specific receptors, the best characterized being low-density lipoprotein-related protein 1 (LRP-1) (106). Those mechanisms, however, have not been elaborately studied in relation to disease mechanisms and causes of neurodegeneration and dementia. In young mice, the clearance ratio of labeled Aβ across the BBB compared to the CSF route is nearly 9:1. However, with aging, LRP-1 transport is reduced by 50% (106). This would suggest a greater role for CSF production and turnover in Aβ clearance in aging brains, although CSF production and turnover, the secondary clearance route for Aβ, has also shown a two- to fourfold decrease in aging, hydrocephalus, and AD (107, 108).
BBB barrier dysfunction and leakage has been suggested in NPH as indicated by increase of CSF albumin ratios in patients (41, 109). In the kaolin-induced hydrocephalus of the aging rat, LRP-1 receptor staining dramatically decreased in both cortical and subcortical microvessels (100, 101). This resembled an accelerated effect of aging in hydrocephalus as the LRP-1 in 12-month-old hydrocephalus rats dropped down to levels seen in the 36-month-old Brown–Norway–Fisher senescent rats when comparing the immunohistochemical findings in kaolin-induced hydrocephalus with the findings of microvessel PCR in the Brown–Norway–Fisher aging rat (100). The findings suggest that Aβ accumulation and Alzheimer’s pathology in chronic hydrocephalus of the elderly may be, at least in part, due to a failure of Aβ clearance, resulting from failure of the BBB transporters of Aβ (101). The decrease in LRP-1 receptor density has, in fact, been recently demonstrated in human AD brains (110). The dramatic change in BBB receptor ratios in both NPH and AD should lead to a net flux of Aβ into the brain (101, 110).
Neurofibrillary tangles (NFTs) are a primary pathological constituent of AD, and the number of NFTs present is well correlated to the severity of the dementia (111). A sequential phosphorylation of Tau protein has been reported as representing morphological “stages” of NFT formation: intraneuronal pre-NFT, intraneuronal NFT, and extraneuronal NFT are each phosphorylated at specific amino acids, constituting specific epitopes of hpTau (112). In the kaolin-model of the aging rat, an increased staining of both intraneuronal pre-NFT-hpTau (hp pT 231 and hp pT 262) and extraneuronal NFT-hpTau (AT 100) were investigated (113). At 6 and 10 weeks post hydrocephalic induction, pT231 staining showed neurons with focal somatic “condensed” increases of staining intensity, and loss of dendritic staining, attributable to hpTau migration from dendrites to the neuron soma (“excess Tau”), as seen in AD (113), while pT 262 did not display any differences. AT 100, the Tau variant that stains extracellular NFT’s, containing substantial filamentous Tau – the last step in the progression of NFTs (113, 114) – showed increased staining in the hippocampus – Fig. 6. The prominent extracellular AT100 staining in the hippocampus raises further evidence towards a reflection of the stereotypical, sequential, and regional, hierarchical distribution of Tau pathology seen in AD (111), in the course of aging kaolin-induced hydrocephalus.
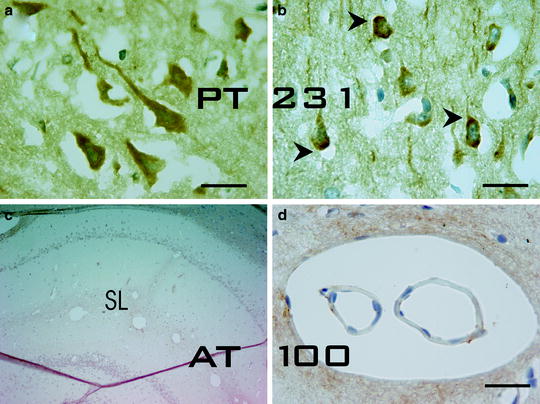
Fig. 6.
Tau Pathology in the kaolin-induced hydrocephalus model of the aging rat. The photomicrograph shows cortical antihyperphosphorylated Tau pt 231 positive staining in a 12-month-old nonhydrocephalic rat (a) and in a hydrocephalic rat 10 weeks after kaolin induction (b). In the control animals, homogeneous age-related staining is seen throughout the soma and the dendrites of neurons, whereas, at 10 weeks, almost all neurons display a more irregular staining pattern with positively stained coarse granules in the soma periphery and at the axon hillock (arrowheads), areas of “excess” Tau accumulation in AD [113]. In the hippocampus of the hydrocephalic rats (c), extracellular AT100 staining was observed at 6 weeks. This antibody specifically recognizes paired helical filament Tau (PHF-Tau). The epitope of this antibody has been shown to contain phosphorylated Serine 121 and phosphorylated Threonine 214 residues (numbering according to human Tau 40) and prominently stains extracellular NFT’s, containing substantial filamentous Tau, the last step in the progression of NFT’s.The AT100-positive staining displayed a punctuate granular pattern in extracellular regions in proximity to hippocampal vessels, and was clearly associated with them (d); bar = 25 µm.
In NPH patients, reports differ regarding Tau levels in the lumbar CSF; both increases (115) and decreases (116, 117) have been observed. Neither study differentiated between regular and hpTau. Agren Wilson and coworkers (52) reported both low hpTau and Aβ levels in the lumbar CSF to be diagnostically relevant for NPH patients. In light of the findings on the Aβ and hpTau found in the kaolin-model, further evidence is needed on how reliably lumbar CSF protein content reflects CSF content as a result from cortical destruction or periventricular damage in dynamically altered CSF circulation of the hydrocephalic brain (100, 118). The different response in the hpTau variants in the kaolin model (119) warrants the study of the specific hpTau variants in human hydrocephalus.
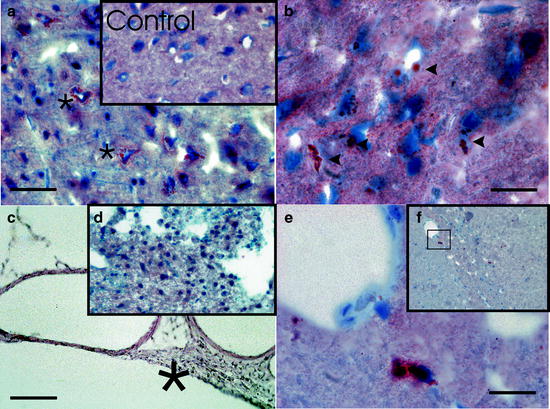
Finally, the third component, the inflammatory oxidative stress response was evidenced by using a marker seen in and around dystrophic neurons and Aβ plaques in advanced AD, i.e., Lipoxygenase 12/15 (LOX12/15), probably being the major source of the oxidative stress (120, 121). LOX12/15 belongs to the family of lipid-peroxidizing enzymes, and has dual-specificity for forming both toxic arachidonic acid metabolites, 12 (S) and 15 (S) Hydroperoxy- eicosatetraenoic acids (12 (S) Hp ETE and 15 (S) Hp ETE) in a ratio of 3:1. In the kaolin-hydrocephalus of the aged rat, using the LOX 12/15-specific antibody as used in the AD-brains (120), immunostaining exhibited morphological findings in cortical dystrophic neurons and microglial-type cells similar to what has been found in AD samples (119, 122). Colocalization of the LOX 12/15 particles with hpTau and Aβ using CY-2 and CY-3 coupled double fluorescence staining added further support to the existence of the pathological cascade (123) in hydrocephalus – Fig. 7.
< div class='tao-gold-member'>
Fig. 7.
Inflammatory Alzheimer-related response pathology in the kaolin-induced hydrocephalus model of the aging rat. Photomicrograph showing cortical 12/15 Lox positive staining in 12-month-old nonhydrocephalic rat (inset, ×40 magnification), and at 6 weeks after induction of kaolin hydrocephalus (a, bar = 50 µm). At 6 weeks of hydrocephalus, abundant positively stained “spots” are seen in and around neurons (asterisks). In controls, only rare and much smaller LOX positive spots are seen around neurons. At higher magnification (b, bar = 25 µm), the Lox-positive staining is intense within neuronal cell bodies and processes in the hydrocephalic rats (arrowheads), as described in Alzheimer’s disease (AD) by Praticó et al. (AJP May 2004, Vol 164, No.5). In the subarachnoid space, however, where the kaolin-related chronic arachnoid inflammation is found (H&E; c, bar: 50 µm), no significant LOX-positive staining is observed (d, ×200 magnification). LOX-positive staining was also seen in cells that morphologically resemble microglia (e, bar = 25 µm). These cells were seen near vessel borders, predominantly observed in the hippocampus stratum lacunosum as seen in the overview (f, ×100 magnification).
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


