, Mohammad Z. Hossain2 and John T. Weber3
(1)
Department of Biology, University of Prince Edward Island, Charlottetown, PE, Canada
(2)
Division of BioMedical Sciences, Faculty of Medicine, Memorial University of Newfoundland, St. John’s, NL, Canada
(3)
School of Pharmacy and Division of BioMedical Sciences, Faculty of Medicine, Memorial University of Newfoundland, St. John’s, NL, Canada
Abstract
Dementia in humans following traumatic brain injury (TBI) has been well documented in clinical populations, either after a single TBI or repeated mild TBI (rMTBI). In most cases, trauma-induced dementia follows a slow, chronic time-course, and in many cases mild injuries accumulate over time. Both single TBI and rMTBI have been modeled experimentally in vivo, most often in rodents, and various treatment strategies have been studied in order to reduce brain damage after injury. Here, we review the recent literature with regard to currently used in vivo models of TBI in animals, and studies conducted with them to investigate the link between TBI and the development of dementia-like pathology, such as that associated with Alzheimer’s disease (AD). We also discuss the potential use of in vitro models of trauma for investigating a link between trauma and dementia.
Key words
Animal modelAlzheimer’s diseaseDementiaMouseRatRepeated injuryTBI1 Introduction
Traumatic brain injury (TBI) is generally defined as an injury to the brain caused by an external physical force, resulting in total or partial functional disability. Motor vehicle accidents are the primary cause of TBIs, while falls, sports, assaults, and gunshot wounds are also major causes of these injuries (1, 2). Globally, the incidence of TBI is reported as approximately 200 in 100,000 individuals with a mortality rate of about 20 per 100,000, making it one of the leading causes of death and disability worldwide (1, 2). In the United Kingdom, 200–300 per 100,000 people are hospitalized each year due to TBI (3), and the incidence is even higher in areas such as South Africa and southern Australia (4, 5). In the United States, TBI is associated with the death of approximately 51,000 people each year and causes long-term disability that affects an estimated 70,000–90,000 individuals annually (6). TBI is particularly prevalent in young individuals, and is the leading cause of mortality in infants (7). In the United Kingdom, TBI may represent as much as 20% of all deaths of individuals in the range of 5–45 years of age (5). In 1999, a National Institutes of Health Consensus Development Panel suggested that as many as 6.5 million individuals may be living with the consequences of TBI in the United States. In Europe, at least 11.5 million people suffer from disabilities related to a TBI (8). Although the total financial burden of TBI is difficult to determine, estimated costs of head injury per year in the United States alone are over $1 billion (9, 10). Given the global prevalence and enormous socioeconomic costs of TBI, it is important to understand the mechanisms that contribute to long-term dysfunction following trauma, so that we may devise appropriate treatment strategies to improve patient outcome.
The pathophysiology of TBI consists of two main phases: a primary (mechanical) phase of damage and a delayed secondary phase of damage. Primary damage occurs at the moment of sudden traumatic insult and includes contusion and laceration, intracranial hemorrhage, and diffuse axonal injury (10–13). Secondary damage includes processes that are initiated at the time of insult, but do not appear clinically for hours, or even days after the insult has occurred. Secondary processes include brain damage due to altered neurochemical mechanisms (e.g. increased glutamate release, generation of free radical species), activation of degradative enzymes (e.g. proteases), swelling (edema), and ischemia (for reviews, see (14–21)). TBI is also further defined by the severity of its symptoms, primarily based on the Glasgow Coma Scale (GCS) (22). Major classifications include severe TBI, which has a high fatality rate, moderate TBI, and mild TBI, which includes concussion and does not necessarily involve a loss of consciousness.
Studies and case reports from Europe in the 1950s and 1960s highlighted the coincidence of brain injury and cerebral deficits in specific human populations, such as dementia pugilistica (DP) in professional boxers (23–25), which results in severe motor and cognitive dysfunctions (26). These observations provided some of the first clues leading to the suggestion that mild, repetitive insults to the head could lead to dementia, most likely caused by neurodegeneration. Dementia is broadly defined as a clinical syndrome of cognitive and functional decline, frequently associated with behavior or personality changes (27). There are several different types of cortical and subcortical degenerative dementias (27, 28). Besides DP, the main cortical dementias include Alzheimer’s disease (AD), vascular dementia, and Diffuse Lewy body dementia (DLBD), whereas subcortical dementias can result from diseases such as Huntington’s, Parkinson’s, syphilis, and AIDS. Some of these diseases are primarily associated with aging (e.g. AD, vascular dementia), some clearly are inherited (e.g. Huntington’s), while others are due to environmental factors, such as infectious agents (syphilis, AIDS).
As with DP, there is growing evidence of a link between TBI and AD, suggesting that this type of neurodegenerative disease can be environmentally triggered. Initial evidence for this relationship was derived from a case report in which a 38-year-old man, who had experienced a TBI 16 years previously, developed early-onset AD (29). Since then, several epidemiological studies have been conducted to investigate this interaction. Although many studies have found no conclusive connection between TBI and AD, many others have demonstrated a definite relationship, and TBI is generally considered a robust risk factor for the future development of AD (for reviews, see (30, 31)).
As can be deduced from the literature, evidence for a link between head injury and the eventual development of cerebral dementia is correlative in nature. What is needed are in-depth studies with experimental models of TBI in order to demonstrate a true existence of a cause-and-effect relationship between trauma and dementia, as well as an understanding of the mechanisms that can lead to dementia following an injury, or repeated injuries, to the head. Although this area of research is largely in its infancy, several experimental approaches used to analyze trauma-induced dementia have been developed in recent years. This review provides an overview of the experimental methods that have been utilized to study the link between TBI and dementia.
2 In Vivo Models of Traumatically Induced Dementia: Single Injury
In addition to cognitive symptoms, dementias such as DP and AD are associated with specific types of neuropathological markers. In fact, AD in humans can only be fully confirmed postmortem via the presence of extracellular senile plaques, which are abnormal amyloid β (Aβ) protein deposits, and abnormal tau protein aggregation in specific brain regions (32). The tau protein is a functional component of the neuronal cytoskeleton of healthy neurons, and is a key component of the neurofibrillary plaques that are seen in both DP and AD (33). As with much basic research aimed at understanding neurodegenerative diseases, one is often shrinking down a large portion of a patient’s lifetime into a few years or less. This can be challenging in one aspect, but beneficial if a disease state can be modeled properly. Any thorough study of the link between TBI and dementia should in theory encompass cognitive assessments of animals, as well as neuropathological evaluations at the conclusion of the study.
Early investigations into the effects of trauma on the development of dementia-like pathology in animals were largely descriptive in nature. In general, these studies utilized trauma devices, which delivered a head injury – usually to rats or mice – and measured cognitive impairment at various time points. These experiments also attempted to replicate clinical findings in human cadavers, in particular the existence of neuropathological markers, such as tau and Aβ protein, in animal brains after trauma (30, 31). Although moderate and severe TBI in humans have been fairly well characterized, satisfactory treatment options have been slow to develop. Recently, increased emphasis has been placed on patients who suffer from single or repeated mild TBI (rMTBI) as this population not only suffers from cognitive deficits, but may also respond better to treatment. Indeed, Sosin et al. (34) pointed out that over a million concussions occur annually in the United States, with a third of those cases resulting from sports-related injuries. Given the large number of new TBI cases worldwide per year, especially mild TBI, the coincidence of TBI and dementia can only increase. As the world population continues to age, however, especially in Western nations, the need for mechanistic studies has greatly increased. Overall, current aims have been to treat injured patients and to predict their recovery outcomes, the assumption being that there is a definite, causal link between TBI and dementia, such as AD.
Animal models of cognitive dysfunction, in particular those that use mammals, are based on the relative conservation of neuroanatomical structure–function relationships between humans and other mammals, such as rodents (35). To this end, multiple tests have been developed with which to discern cognitive deficits in rodents, including the Morris water maze (MWM), object recognition, fear conditioning, and conditioned taste aversion (35). These paradigms can be particularly useful after mild TBI, where impairment between groups may be more subtle. That being said, Nakayama et al. (36) believe that our current use of animal models for dementia may be hampered by the fact that they may die before the signs of neurodegenerative diseases are manifested. In addition, any deficits in cognitive tests in rodents subjected to experimental TBI may be due to dementia, or to the more immediate effects of the trauma itself. Therefore, the time it takes to observe cognitive deficits in animals, or the notion that cognitive deficits may worsen with time after injury, are important characteristics to take into account in these animal models.
Several experimental devices have been developed, which deliver reliable and reproducible brain injuries to rodents. The two most commonly used models in studies of traumatically induced dementia are lateral fluid percussion injury (LFPI) and controlled cortical impact (CCI). Injury severity can be easily adjusted with these in vivo TBI models, in order to study mild, moderate, or severe levels of injury. To eliminate any confounding effects from surgery or anesthesia, “sham” animals are used as comparative controls, in that they undergo the same procedures as injured animals without the actual head injury.
LFPI is an experimental model of concussive TBI used in rodents (Fig. 1). In this model, a pendulum strikes a piston sending a brief percussion pulse of saline into the extradural space of the closed cranial cavity causing a quick displacement and deformation of the brain, mimicking concussive injury. The severity of the injury is determined by adjusting the height of the pendulum. LFPI results in focal as well as diffuse brain injury consisting of a cortical contusion at the impact site and cell death and axonal damage in the hippocampus, thalamus, and cortex (37–40). LFPI is widely used since it reproduces many pathophysiological features of human TBI including breakdown of the blood-brain barrier, neuronal cell loss, gliosis, altered ionic homeostasis, and long-term alterations in cognitive and motor function (37, 38, 40). LFPI has been extensively characterized and widely used in rats, and more recently in mice (39, 41). Although data generated in rats make for easier comparisons to the majority of previous studies in this animal, the use of mice to study TBI allows investigators to exploit transgenic and knockout mouse technology to directly test the role of specific genes or gene mutations in TBI.
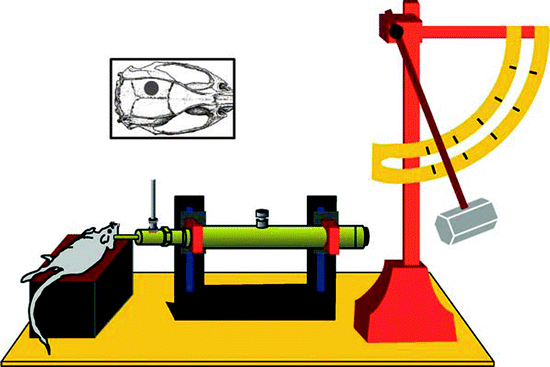
Fig. 1.
Lateral fluid percussion injury (LFPI) model. Representative placement and site of originally described lateral (parasagittal) fluid percussion brain injury (inset) and schematic diagram of a fluid percussion brain injury device. A pendulum from a known height impacts the piston of a saline-filled reservoir, forcing a brief fluid bolus into the sealed cranial cavity (Reproduced from Thompson et al., 2005 with permission from Mary Ann Liebert, Inc.).
CCI is an in vivo model of TBI (Fig. 2), which produces a highly quantifiable impact to the brain or skull (13, 42). As with LFPI, animals generally undergo a craniotomy, and the severity of injury is determined by the impact velocity and duration of the impactor, which causes displacement of the brain of various levels. This model can also be used in the absence of craniotomy, which produces a closed head impact model, which more closely mimics TBI in the human population. Qualitatively, this model produces a similar pattern of injury throughout the brain when compared to LFPI. Some major advantages of the model are that the impact produced by CCI can be scaled across species due to its highly quantifiable nature (43), and it can be used to produce injury in several different areas of the cortex.

Fig. 2.
Controlled cortical impact (CCI) model. Animals are anesthetized and are injured with an impactor either with or without craniotomy in a specific region of the skull. The severity of injury is determined by the impact velocity and duration of the impactor tip (photo supplied courtesy of Prof. Nikolaus Plesnila, Royal College of Surgeons in Ireland).
Using LFPI, Pierce et al. (44) showed increased amyloid precursor protein (APP), which is the protein from which Aβ protein is produced, in rats after injury. However, no increase in Aβ was noted. In a follow-up study, Pierce et al. (45) observed cognitive deficits for up to a year after severe LFPI. Masumura et al. (46) showed no changes in APP or Aβ after LFPI in the rat, whereas Murakami et al. (47) demonstrated increased APP in the cerebral cortex and hippocampus, but no increase in Aβ deposition. Ciallella et al. (48) also showed increased APP, but no Aβ increase using the CCI model in rats. In another study using CCI, Blasko et al. (49) found increases in the activity of β-secretase after injury, which is the first protease that cleaves APP, followed by the action of γ-secretase, which then leads to Aβ production. The activity of β-secretase increased transiently for 2 days after injury, but decreased back to basal levels by 7 days post-injury. Overall, these nontransgenic animal studies of TBI have failed to demonstrate a rapid increase in Aβ in the brain after injury.
The weight-drop model, which produces vertical acceleration movement of the head, has also been used to analyze cognitive performance after injury, with interesting results. This model consists of a mass free falling a known distance over a friction free guide rod and onto an impactor device, which is positioned on the head of the animal. Milman et al. (50) administered single, mild injuries to mice, and measured their cognitive abilities at 7, 30, 60, and 90 days post-injury using the swim T-maze and passive avoidance paradigms. They found that injured mice performed significantly worse than uninjured mice at day 30 only, indicating that the onset of cognitive impairment was slow, and that recovery from injury was possible. Slow onset of cognitive dysfunction could be indicative of dementia, but a recovery from such symptoms is not common in the human population.
To date, most of the previous studies have focused on the development of dementia, or specifically AD-related dementia, after moderate or severe levels of injury. Recent reviews have highlighted the advances being made in the generation of animal models of traumatically induced AD (31, 35). The focus on AD is understandable, as it accounts for more than half of all age-related dementias in modern society (51). However, the benefits of using transgenic animal models of dementia, such as AD (e.g. reduced time until disease onset, clear evidence of neuropathology) must be weighed against the relatively small percentage of cases with an obvious genetic link. For example, almost 90% of all AD cases are believed to be sporadic (52). Nonetheless, these studies provide insights into some of the potential pathological signs of AD that occur after TBI.
Recently, Brody and Holtzman (53) investigated the use of various MWM platform search strategies in wild-type mice, and in transgenic mice that overexpress a mutant human form of APP, before and after a single, moderate injury using the CCI device. Uninjured transgenic mice performed worse on the MWM (i.e. reduced reliance on spatial strategies) than wild-type animals on day 1, but equally as well by day 3. After TBI, however, the transgenic mice demonstrated poor strategy use as compared to injured wild-type mice, and the authors believe that this impairment provides direct comparisons to human TBI patients. Importantly, they did not find any Aβ deposition in transgenic mice, with or without injury; the question remains, however, as to whether Aβ does not play a role in traumatically induced dementia, or if this lack of deposition is attributed to the long-term progression of AD in humans.
Nakagawa et al. (54) also used a transgenic mouse model in which deposition of Aβ accumulates by 6 months of age. Both transgenic mice and wild-type controls were subjected to CCI, or remained uninjured, and brains were examined for Aβ deposition after 2, 5, or 8 months. Interestingly, TBI caused massive hippocampal neuron loss, which occurred concomitantly with a large, brief increase in Aβ, as well as decreased overall Aβ deposition, in older transgenic mice. The authors proposed a “two-hit” hypothesis in an attempt to explain their findings, and those of other groups who have found little or no evidence linking TBI and the development of dementia, particularly AD. In their hypothesis, TBI constitutes the second-hit, in which damage is manifested only after the first hit: high concentrations of Aβ.
In some of the earliest studies analyzing the effects of trauma on the development of AD-like pathology, Murai et al. (55) failed to demonstrate increased Aβ levels after injury induced by CCI in a transgenic mouse model that overexpresses human APP by twofold. In addition, injured animals showed similar motor and cognitive deficits as wild-type mice. However, Smith et al. (56) showed transient increased Aβ levels in the hippocampus of transgenic animals, which overexpress human APP by tenfold. This finding also correlated with a greater loss of neurons in the CA3 region of the hippocampus than wild-type mice. However, the formation of amyloid plaques occurred no faster than in injured wild-type mice. As with the studies in nontransgenic animals, rapid Aβ deposition has not been demonstrated as it has been in human studies (57–60). Also, an increase in the severity of injury does not seem to lead to an increase in Aβ deposits (30), and in some cases a higher level of injury may reduce Aβ deposition. This may be because more neurons containing Aβ are dying and being cleared from the brain after injury, but again, this observation does not correlate with findings in humans, where Aβ levels seem to increase with injury severity (58), as does the risk of developing AD (61).
DLBD in humans has been associated with an accumulation of neurofilament (NF)-rich inclusions. Although the mechanisms behind this formation are unclear, the fact that NFs are critical for proper neuronal function indicates that these inclusions could decrease cell survival. To this end, Nakamura et al. (62) subjected transgenic mice that develop Lewy-body-like inclusions, and their wild-type littermates, to single TBI using the CCI device, and evaluated their neuromotor function postinjury. Although injured wild-type mice showed dysfunction postinjury as compared to uninjured wild-type mice, injured transgenic mice exhibited motor difficulties for significantly longer periods of time posttrauma than all other groups. Therefore, TBI appeared to exacerbate the effects of dementia-like pathology in this model.
There is potential to analyze dementia-like pathology in nonrodent species. For example, using an ovine head impact model, Van Den Heuvel et al. (31) demonstrated a rapid increase in APP mRNA expression after injury. Smith et al. (64) also showed accumulation of Aβ in the brains of pigs subjected to rotational head injury, as early as 3 days after trauma. A study using rotational head injury in rabbits demonstrated a slight increase in Aβ after injury and a redistribution of NFs (65). Head et al. (66) found hallmarks of AD in aged, but not young cats, indicating that the cat may also be a good model for traumatically induced dementia. Drawbacks of using cats, however, include their long life-span – the “aged” cats used were 16 years of age or older – and the scarcity of studies using cats for acceleration–deceleration injury research. However, a longer life-span may also be a positive factor that would more closely mimic the human lifespan than rodents, which generally only live 1–2 years. In addition, you would need dedicated researchers and resources to carry out such long-term studies analyzing the mechanisms and possible therapeutic interventions to inhibit dementia after TBI.
3 In Vivo Models of Traumatically Induced Dementia: Repetitive Injury
Considerably less work has been conducted regarding the potential relationship between rMTBI and dementia. One of the potential problems with mimicking rMTBI is that, often, many years must pass before the onset of dementia in human patients. As a result, epidemiological data from human populations has been slow to accumulate; in this case, proper animal models could help decipher the mechanisms by which dementia may be triggered by repetitive brain injury.
Several lines of evidence suggest that rMTBI may have cumulative effects, leading to dementia-like neuropathology. In a recent review (67), rMTBI was defined as an initial mechanical insult to the head, followed by another mechanical insult to the head of the same or different magnitude. The development of animal models for the study of rMTBI has drawn largely on the findings after single moderate or severe injury. In most cases, rats or mice are subjected to one or multiple mild injuries, and both behavioral (e.g. MWM) and histopathological (e.g. silver stain) outcomes are measured. These studies often fall in one of two categories: the effect of rMTBI on normal mice (e.g. C57BL/6 strain) or the effect of rMTBI on transgenic mice (e.g. Tg2576 AD transgenic line).
Initial studies utilized normal mice and simple injury paradigms. Laurer et al. (68) administered zero, one, or two injuries (spaced 24 h apart) to adult mice, and found marked differences among the experimental groups. Although they did not find cognitive impairment in all three groups using the MWM, they did find exacerbated functional and behavioral impairment in the single MTBI mice. Moreover, whereas this impairment lasted for 3 days in the single MTBI animals, these deficits were found in rMTBI up to 7 days postinjury. Their work highlighted the importance of the interinjury interval, a parameter that can easily be adjusted bothin vivo and in vitro. Similarly, Creeley et al. (69) utilized the weight-drop paradigm, in which 7-week-old mice were subjected to either three impacts, or remained uninjured. Interestingly, mice that experienced rMTBI were significantly slower to regain consciousness than uninjured mice. Spatial learning was also found to be markedly impaired in these mice using the MWM, possibly due to contrecoup brain injury, which is an injury occurring opposite to, or distant from, the sight of impact.
Later studies compared the effects of either multiple injuries or a variable interinjury interval. Longhi et al. (70) compared the cognitive functioning in mice receiving either zero, one, or two injuries (spaced either 3, 5, or 7 days apart), and found that, whereas mice injured twice after 3 or 5 days had significantly increased cognitive impairment compared to sham or single-injury mice, this impairment was not seen in mice injured twice 7 days apart. Their findings highlighted the fact that the interinjury “window” does not extend indefinitely after two injuries; what remains unknown is if that vulnerability would be increased after three or more injuries.
Similar to the numerous studies measuring the effects of a single trauma on the development of dementia, the progression of AD-like impairment has been a major focus of rMTBI research in vivo. As mentioned previously, the development of abnormal tau protein pathology is a potential molecular link between TBI and dementia. Interestingly, the human brain seems to be more resistant against tau pathology after injury. Yoshiyama et al. (71) used a robust injury paradigm in an attempt to model human DP in transgenic mice expressing the shortest human tau isoform (T44). Mice were subjected to four injuries a day, once a week, for 4 weeks, resulting in each mouse receiving a total of 16 injuries, and, surprisingly, they could find only one mouse that displayed pathology of DP at 9 months of age. Partly for this reason, the vast majority of animal studies have focused on the deposition of Aβ, or the intracellular processing of APP. Interestingly, deposition of Aβ has not been observed in the majority of nontransgenic animal studies after trauma (30, 68), and, as a result, many of the current models used to investigate traumatic dementia are derived from transgenic rodents that were originally created to investigate AD. For example, the transgenic mouse Tg2576, which is characterized by AD-like amyloidosis by 9 months of age, has been used in several investigations of rMTBI, and has become a popular animal model for traumatically induced dementia.
In contrast to the work of Brody and Holtzman (53) described previously for single injury, Uryu et al. (72) found that Tg2576 transgenic mice subjected to repeated, but not to single MTBI, displayed both cognitive deficits and Aβ deposition. As shown in Fig. 3, Aβ deposition did not occur in these mice at either 9 or 16 weeks post sham injury. In contrast, brain slices from Tg2576 mice that underwent rMTBI showed marked Aβ deposition (in the form of senile plaques) at 16 weeks postinjury. This finding highlights two key points. First, the transgenic background alone was not sufficient to induce Aβ deposition in these aged mice, thereby corroborating the “two-hit” hypothesis proposed by Nakagawa et al. (54). Second, the appearance of senile plaques followed a delayed time-scale, which is not surprising, as dementia is often manifested in humans long after TBI.
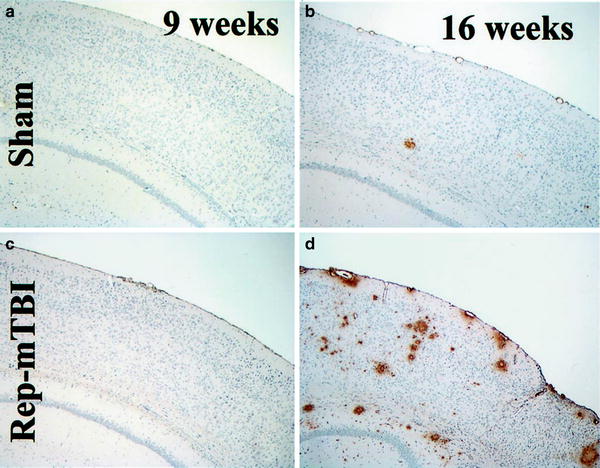
Fig. 3.
Amyloid deposition in Tg2576 mice with sham or repeated mild traumatic brain injury (rMTBI) (b, d) with 4G8 immunohistochemistry at 9 (a, c) and 16 (b, d) weeks after mTBI. Senile plaques increased in an age-dependent manner in both sham and injured mice, but the largest number of Aβ-positive plaques are seen in the 16-week Rep-mTBI mice (d) (Modified from Uryu et al. (2002). Reprinted with permission from the Society for Neuroscience, 2002).
Increased incidence of dementia in humans has repeatedly been associated with increased age (73–76), and recent evidence links aging with the overproduction of free radicals via oxidative stress (21). TBI is also known to dramatically increase free radicals and reactive oxygen species (21, 77). Repetitive, but not single MTBI, has been previously shown to increase oxidative stress in Tg2576 mice (72), which was reduced by supplementing vitamin E, a known antioxidant, into the rodent chow (78). Therefore, oxidative stress may be one of the major contributing factors leading to the development of neurodegenerative disease following TBI.
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


